 |
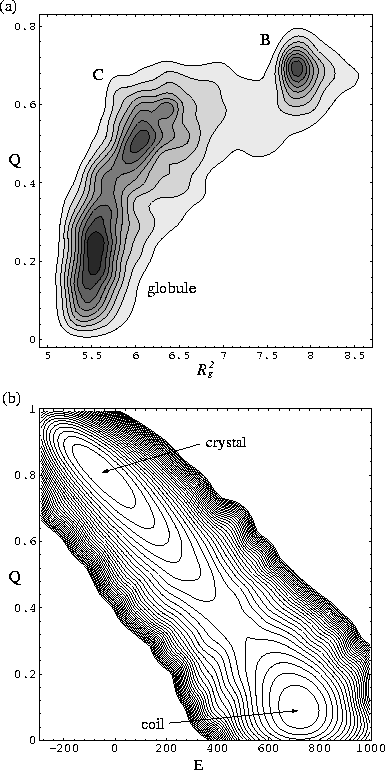
At temperatures in the transition region, as for the solid-solid transitions,
dynamic coexistence of the ordered and disordered states is seen, leading to
multimodal probability distributions (and free energy barriers) [Figure 7].
In the example shown in Figure 7a three states are seen.
Structures based on the global minima B and C give rise to the two ordered states.
The low value of Q (![]() ) for the state associated with structure C is a
reflection of the considerable disorder that can be present in the solid
state at Tm.
The peak with the lowest Q value is due to the disordered globule and
an example of a polymer configuration that contributes to this peak is given in
Figure 3c.
) for the state associated with structure C is a
reflection of the considerable disorder that can be present in the solid
state at Tm.
The peak with the lowest Q value is due to the disordered globule and
an example of a polymer configuration that contributes to this peak is given in
Figure 3c.
 |
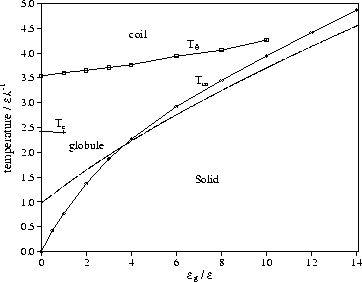
The effects of stiffness on this order-disorder transition can be seen from
Figures 6 and 8.
The temperature of the transition increases with stiffness, and
at larger values of ![]() this increase is a little slower than linear.
Accompanying this change is an increase in the height of the heat capacity peak, and
therefore the latent heat of the transition.
This increasing energy gap between the ordered and disordered states is a
result of the increasing energetic penalty for the larger number of gauche
bonds associated with the disordered state.
As
this increase is a little slower than linear.
Accompanying this change is an increase in the height of the heat capacity peak, and
therefore the latent heat of the transition.
This increasing energy gap between the ordered and disordered states is a
result of the increasing energetic penalty for the larger number of gauche
bonds associated with the disordered state.
As ![]() , the dependence of the energy gap,
, the dependence of the energy gap, ![]() , on the stiffness is
one of the main causes of the increase in Tm.
This effect is reinforced by the changes in the entropy difference between ordered and
disordered states,
, on the stiffness is
one of the main causes of the increase in Tm.
This effect is reinforced by the changes in the entropy difference between ordered and
disordered states, ![]() :as
:as ![]() increases the number of gauche bonds decreases, thus lowering the number of
configurations contributing to the disordered state.
increases the number of gauche bonds decreases, thus lowering the number of
configurations contributing to the disordered state.
 |
This behaviour is similar to simple liquids where decreasing the range of the potential leads to an increasing energy gap between solid and liquid because of the increasing energetic penalty for the disorder associated with the dispersion of nearest-neighbour distances in the liquid, thus playing an important role in the destabilization of the liquid phase.[34,35]
The respective roles of energy and entropy in the increase of Tm with stiffness
can be investigated more quantitatively by approximating the free energy of the disordered state by that
for the ideal coil. This is a reasonable approximation, since
the ideal expression for ng, ![]() ,fits the simulation values for the coil, and for the globule, fairly closely.
The free energy of an ideal coil Aideal is
,fits the simulation values for the coil, and for the globule, fairly closely.
The free energy of an ideal coil Aideal is
| |
(7) |
Use of Equation (7) also allows us to calculate a value of Tm if
we approximate the free energy of the solid by only its energetic component
[Equation (5)].
This simple calculation gives surprisingly good agreement with the simulation
data [Figure 8], especially at larger ![]() .
It breaks down at low
.
It breaks down at low ![]() , e.g. the prediction of a non-zero value of Tm
at
, e.g. the prediction of a non-zero value of Tm
at ![]() =0, because Equation (7) is the free energy of a coil in
the absence of any interactions;
for the dense polymers at low
=0, because Equation (7) is the free energy of a coil in
the absence of any interactions;
for the dense polymers at low ![]() and near to Tm, the polymer-polymer
contacts make a significant contribution to the energy of the disordered state.
The number of polymer-polymer contacts in the disordered polymer decreases significantly
with increasing temperature, because of the larger entropy of less dense configurations.
Therefore, Equation (7) becomes a better approximation to the free energy
at higher temperatures, and thus provides a better description
of melting at the higher temperatures relevant for larger
and near to Tm, the polymer-polymer
contacts make a significant contribution to the energy of the disordered state.
The number of polymer-polymer contacts in the disordered polymer decreases significantly
with increasing temperature, because of the larger entropy of less dense configurations.
Therefore, Equation (7) becomes a better approximation to the free energy
at higher temperatures, and thus provides a better description
of melting at the higher temperatures relevant for larger ![]() .
.
Our simulation results for Tm are also in qualitative agreement with theoretical results in
which a more sophisticated treatment of the globule[15] gives the correct
behaviour for Tm at low ![]() .
However, a drawback of the description of Doniach et al. is that the free energy
of the coil per polymer unit is nonzero in the limit of large
.
However, a drawback of the description of Doniach et al. is that the free energy
of the coil per polymer unit is nonzero in the limit of large ![]() ,
causing Tm to reach an asymptotic value,
rather than continuing to increase with
,
causing Tm to reach an asymptotic value,
rather than continuing to increase with ![]() .
.
The increase in ![]() with stiffness also has an effect on the coexistence of
ordered and disordered states.
At large values of
with stiffness also has an effect on the coexistence of
ordered and disordered states.
At large values of ![]() there is multimodality in the
probability distribution for the energy as well as for the order parameter,
because of larger free energy barriers between the states.
In the example shown in Figure 7b there is a free energy barrier of 3.24 kT
for passing from the crystal to the coil.
there is multimodality in the
probability distribution for the energy as well as for the order parameter,
because of larger free energy barriers between the states.
In the example shown in Figure 7b there is a free energy barrier of 3.24 kT
for passing from the crystal to the coil.
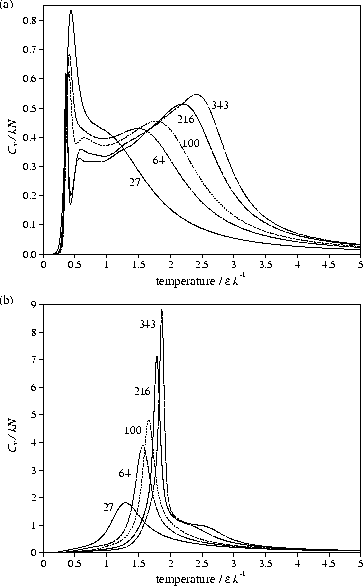
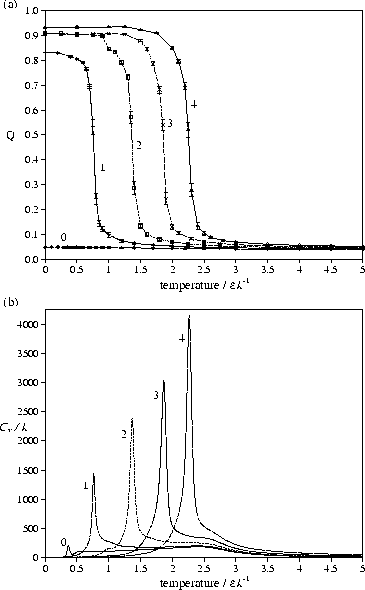
A consideration of the effect of size on the transition
shows that as the polymer becomes longer the melting point becomes higher,
and the transition becomes sharper with an increasing latent heat per monomer [Figure 9b].
This is consistent with the transition being the finite-size analogue of a first-order phase transition.
Tm increases with size because the effect of the surface term (the N2/3 term) in the
energy of a crystallite [Equation (5)] diminishes with size.
Moreover, as the coefficient of the surface term increases with ![]() (the higher aspect
ratio crystallites have a larger surface area)
the effects of size on Tm are more pronounced at larger
(the higher aspect
ratio crystallites have a larger surface area)
the effects of size on Tm are more pronounced at larger ![]() .
.
At ![]() =0 the behaviour is qualitatively different because
there is no energy difference between the orientationally ordered and disordered forms.
As there are far fewer states that possess orientational order, they
are never thermodynamically favoured and so there is no orientational order-disorder
transition and Q always has a low value [Figure 6a].
Despite this, the polymers we studied do have a low temperature heat capacity
peak at
=0 the behaviour is qualitatively different because
there is no energy difference between the orientationally ordered and disordered forms.
As there are far fewer states that possess orientational order, they
are never thermodynamically favoured and so there is no orientational order-disorder
transition and Q always has a low value [Figure 6a].
Despite this, the polymers we studied do have a low temperature heat capacity
peak at ![]() [Figure 9a].
This feature stems from a transition between the maximally compact cuboidal global minimum and
a more spherical dense globule, and gives rise to bimodality in the
canonical probability distribution of the energy.
However, the latent heat per atom for the transition decreases with increasing size,
indicating that the transition is not tending to a first-order phase transition in the bulk limit.
Furthermore, the form of the heat capacity can be very different for sizes
at which it is not possible to form complete cuboids.
[Figure 9a].
This feature stems from a transition between the maximally compact cuboidal global minimum and
a more spherical dense globule, and gives rise to bimodality in the
canonical probability distribution of the energy.
However, the latent heat per atom for the transition decreases with increasing size,
indicating that the transition is not tending to a first-order phase transition in the bulk limit.
Furthermore, the form of the heat capacity can be very different for sizes
at which it is not possible to form complete cuboids.
The order-disorder transition observed by Zhou et al. was also for a fully flexible polymer.[10] However, as the model used was off-lattice, unlike in our model, there is the possibility of a first-order transition associated with the condensation of the monomers onto a lattice.