In Table I the properties of the global minima for N=100 are given for
the range of ![]() we consider in this study, and examples of these
minima are illustrated in Figure 2.
The shapes of the global minima agree well with that expected based on the analysis
of section IIA.
In the final column of Table I we have given the value of the stiffness
for which a crystallite with the aspect ratio of the global minimum is
expected to be lowest in energy based on Equation (4).
This value generally lies in the middle of the range for which the structure
is the global minimum.
Most of the global minima have a degeneracy associated with the different
possible ways of folding the chain back and forth.
This degeneracy decreases as the chains become stiffer and the crystallites become more extended.
For
we consider in this study, and examples of these
minima are illustrated in Figure 2.
The shapes of the global minima agree well with that expected based on the analysis
of section IIA.
In the final column of Table I we have given the value of the stiffness
for which a crystallite with the aspect ratio of the global minimum is
expected to be lowest in energy based on Equation (4).
This value generally lies in the middle of the range for which the structure
is the global minimum.
Most of the global minima have a degeneracy associated with the different
possible ways of folding the chain back and forth.
This degeneracy decreases as the chains become stiffer and the crystallites become more extended.
For ![]() there is no constraint on ng, and so the global minimum has
a much larger degeneracy, and the majority of the isomers of the global minimum
have no orientational order.
there is no constraint on ng, and so the global minimum has
a much larger degeneracy, and the majority of the isomers of the global minimum
have no orientational order.
| npp | ng | |||||
| A | 136 | 38 | 0.000 | 0.375 | 1.118 | 0.059 |
| B | 133 | 30 | 0.375 | 0.500 | 1.563 | 0.281 |
| C | 129 | 22 | 0.500 | 0.833 | 2.406 | 0.703 |
| D | 124 | 16 | 0.833 | 2.167 | 3.704 | 1.352 |
| E | 111 | 10 | 2.167 | 3.500 | 6.804 | 2.902 |
| F | 97 | 6 | 3.500 | 12.000 | 12.500 | 5.750 |
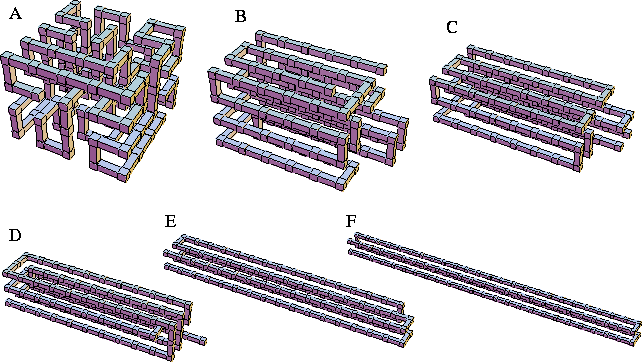 |
The global minimum is the free energy global minimum at zero temperature.
However, as the temperature is increased it will become favourable to introduce
defects into the structures.
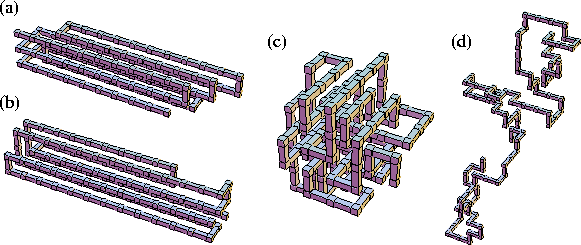
One of the mechanisms we commonly observed was through fluctuations
in the lengths of the folds, moving in and out like the slide of a trombone.
An example of such a structure is shown in Figure 3a.
The generation of this type of defect
is especially common at larger values of
![]() since it does not involve an increase of ng.
since it does not involve an increase of ng.
 |
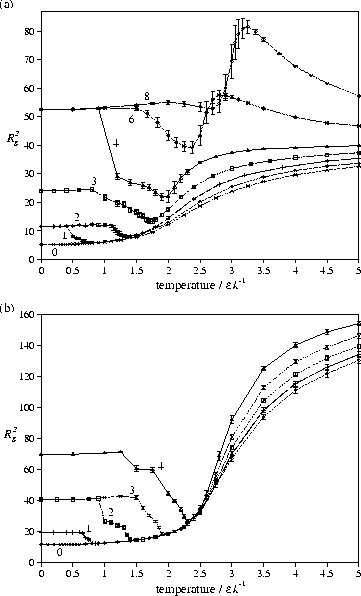
The dependency of the degeneracy, and therefore the entropy,
on the aspect ratio of the crystallites raises the possibility of transitions
to crystallites with a smaller aspect ratio as the temperature is increased.
Indeed, this is usually what is seen and leads to the decrease of Rg2 with temperature
that is observed before melting [Figure 4].
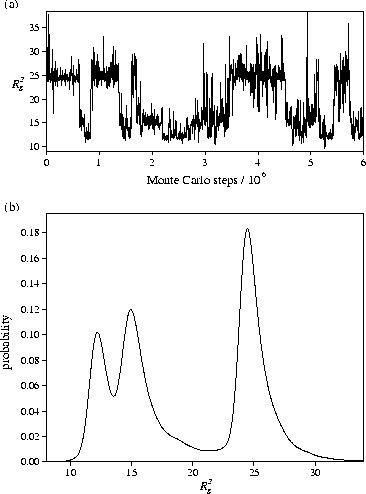
At the centre of the transition, where the free energy of each crystallite is equal,
ideally, the polymer would be seen to oscillate between the two forms [Figure 5a]
during the simulation spending equal time in each.
This `dynamic coexistence' of structures leads to a bimodal
(or even multimodal if more than two forms are stable) distribution of
Rg2 [Figure 5b].
Since the Landau free energy is given by ![]() , where A is the
Helmholtz free energy and pq(q) the canonical probability distribution for an order
parameter q, the multimodality in Rg2 implies that there
are free energy barriers between the different crystallites.
In the example shown in Figure 5, the peak with the highest value of Rg2
corresponds to structures similar to the global minimum D,
and the peak with the lowest value of Rg2 have structures similar to C.
The other peak consists of structures that have eight aligned stems, either in
a
, where A is the
Helmholtz free energy and pq(q) the canonical probability distribution for an order
parameter q, the multimodality in Rg2 implies that there
are free energy barriers between the different crystallites.
In the example shown in Figure 5, the peak with the highest value of Rg2
corresponds to structures similar to the global minimum D,
and the peak with the lowest value of Rg2 have structures similar to C.
The other peak consists of structures that have eight aligned stems, either in
a ![]() array or a square array with one corner unoccupied [Figure 3b].
array or a square array with one corner unoccupied [Figure 3b].
 |
However, isomerization between the different forms often requires a large-scale change in structure,
which is particularly difficult if these transitions occur at low temperature, where
the free energy barriers to transitions are largest.
With simulation techniques that only apply local moves, transitions between crystallites
would be effectively impossible to observe
and even with a technique such as configurational-bias Monte Carlo, where global moves are possible,
transitions may be rare, particularly for the larger polymers in this study.
This possible lack of ergodicity on the simulation time scales can lead occasionally to the abrupt
jumps seen in Rg2 (e.g. for N=100 at ![]() =4
=4![]() and T=1
and T=1![]() ,and for N=343 at
,and for N=343 at ![]() =2
=2![]() and T=0.95
and T=0.95![]() )
rather than the smoother transition that would
be expected if equilibrium values of Rg2 were obtained.
)
rather than the smoother transition that would
be expected if equilibrium values of Rg2 were obtained.
 |
The differences in energy and entropy between crystallites of different aspect ratio are due to surface effects and so scale less than linearly with size. Therefore, these solid-solid transitions are not finite-size analogues of bulk first-order phase transitions.
Interestingly, this coexistence of polymers with different cuboidal shapes but the same basic structure bears some resemblance to the coexistence of cuboidal sodium chloride clusters that has recently been observed experimentally.[33] All the clusters have the rock-salt structure but the cuboids have different dimensions.