In this section we study the escape of the BzAr ![]() cluster from particular regions
of configuration space and compare these to the Monte Carlo results. We start MD
trajectories from the lowest known one-sided and two-sided structures in each
case. We always use the final configuration at a given energy as the starting
point for the next trajectory by rescaling the velocities to give a new kinetic
energy. We loop over trajectories each of 10ns length separated by 1mh in total energy
with the relative
integration accuracy set to
cluster from particular regions
of configuration space and compare these to the Monte Carlo results. We start MD
trajectories from the lowest known one-sided and two-sided structures in each
case. We always use the final configuration at a given energy as the starting
point for the next trajectory by rescaling the velocities to give a new kinetic
energy. We loop over trajectories each of 10ns length separated by 1mh in total energy
with the relative
integration accuracy set to ![]() for the conservation of
energy. Fig. 16 shows the microcanonical caloric
curves (T vs.
for the conservation of
energy. Fig. 16 shows the microcanonical caloric
curves (T vs. ![]() ) produced by these simulations.
Here the calculated kinetic temperature (Eq. 15) is plotted against
the total energy of the trajectory.
) produced by these simulations.
Here the calculated kinetic temperature (Eq. 15) is plotted against
the total energy of the trajectory.
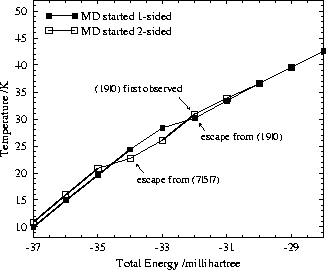
All the kinks in the caloric curves shown in Fig. 16 are symptomatic of escape from certain regions of configuration space, and therefore correspond to non-ergodicity. These escapes are actually of significance in finite time-scale experiments.
We find that an average kinetic temperature of about 23K is needed to escape
from the (7|5|7) starting structure on the time scale of our simulations.
This corresponds to 5mh (1100cm ![]() ) total energy above the -39mh energy
of the (7|5|7) minimum. Consequently, the first
kink in the two-sided curve in Fig. 16 is assigned to escape from
the (7|5|7) minimum. We expect that this kink corresponds to the `melting'
point Fried and Mukamel [4] found in their studies of
) total energy above the -39mh energy
of the (7|5|7) minimum. Consequently, the first
kink in the two-sided curve in Fig. 16 is assigned to escape from
the (7|5|7) minimum. We expect that this kink corresponds to the `melting'
point Fried and Mukamel [4] found in their studies of
![]() and
and ![]() at about 20K. As in earlier work we
considered the Lindemann criterion [4, 46, 51],
i.e. the relative rms
fluctuation of the Van der Waals bond lengths. We observed the same clear
indication of a `melting' process due to the Lindemann criterion as seen by
Fried and Mukamel in their studies and in contrast to the heat capacity results
shown in Fig. 14. However, for all other kinks in the caloric
curve the Lindemann criterion does not appear to be meaningful and we do not
consider it further in the present work.
at about 20K. As in earlier work we
considered the Lindemann criterion [4, 46, 51],
i.e. the relative rms
fluctuation of the Van der Waals bond lengths. We observed the same clear
indication of a `melting' process due to the Lindemann criterion as seen by
Fried and Mukamel in their studies and in contrast to the heat capacity results
shown in Fig. 14. However, for all other kinks in the caloric
curve the Lindemann criterion does not appear to be meaningful and we do not
consider it further in the present work.
The experimentally more important threshold between the one-sided and the
two-sided regime appears at higher energy. At a kinetic temperature of about 30K
(corresponding to 7mh or
1500cm ![]() ) the (19|0) structure changes for the first time to one which is
two-sided in our simulation. This energy difference is equivalent to about
fifty times the energy gap of 0.14mh (31cm
) the (19|0) structure changes for the first time to one which is
two-sided in our simulation. This energy difference is equivalent to about
fifty times the energy gap of 0.14mh (31cm ![]() )
between the lowest lying one-sided and two-sided minima.
The two-sided trajectory consistently becomes one-sided first at about 30K on our
time-scale.
It is noteworthy that both regimes are entropically important at higher temperatures.
Both one-sided and two-sided structures are visited in all of
our simulations above 30K.
These results confirm the expectations from Fried and Mukamel's [4]
studies of
)
between the lowest lying one-sided and two-sided minima.
The two-sided trajectory consistently becomes one-sided first at about 30K on our
time-scale.
It is noteworthy that both regimes are entropically important at higher temperatures.
Both one-sided and two-sided structures are visited in all of
our simulations above 30K.
These results confirm the expectations from Fried and Mukamel's [4]
studies of ![]() and
and ![]() but disagree with the observations
of Adams and Stratt [7] for BzAr
but disagree with the observations
of Adams and Stratt [7] for BzAr ![]() . In the latter MD studies
ring crossing was not observed up to 30K.
We observe side-crossing at temperatures below 30K.
This discrepancy is rather surprising since we use a potential parameter
set which favours Ar-Ar binding compared to the OBJ-like parameters that Adams and
Stratt and Fried and Mukamel used (§iiiA).
However, our
results support the more recent suggestion of Adams and
Stratt that below about 30K the benzene chromophore might be
kinetically trapped in a surface state for significant periods of
time. [8]
. In the latter MD studies
ring crossing was not observed up to 30K.
We observe side-crossing at temperatures below 30K.
This discrepancy is rather surprising since we use a potential parameter
set which favours Ar-Ar binding compared to the OBJ-like parameters that Adams and
Stratt and Fried and Mukamel used (§iiiA).
However, our
results support the more recent suggestion of Adams and
Stratt that below about 30K the benzene chromophore might be
kinetically trapped in a surface state for significant periods of
time. [8]
It is very difficult to decide what time scales are appropriate in the
related experiments [5, 4, 7].
We do not observe a one-sided/two-sided transition
during the 10ns simulation time at 33mh, i.e. 6mh (1300cm ![]() ) above the lowest
minimum. This energy is slightly below our
first estimate of the barrier height. However, if we integrate a longer
trajectory (43ns) we find first one or two Ar atoms occasionally migrating to
the initially non-occupied side but coming back to the main Ar cluster on a
very short time scale. After about 26ns fully `solvated' two-sided structures
occur but it also happens quite often that all Ar atoms come together on one
side again. We believe the one-sided/two-sided transition barrier is very close
to this energy, and it is noteworthy that MD provided a good estimate of the
transition barrier in BzAr
) above the lowest
minimum. This energy is slightly below our
first estimate of the barrier height. However, if we integrate a longer
trajectory (43ns) we find first one or two Ar atoms occasionally migrating to
the initially non-occupied side but coming back to the main Ar cluster on a
very short time scale. After about 26ns fully `solvated' two-sided structures
occur but it also happens quite often that all Ar atoms come together on one
side again. We believe the one-sided/two-sided transition barrier is very close
to this energy, and it is noteworthy that MD provided a good estimate of the
transition barrier in BzAr ![]() (Fig. 15).
(Fig. 15).
A more detailed study of the above feature would involve calculating the rearrangement pathways and the corresponding rate constants. However, we choose not to persue this point further in the present paper.
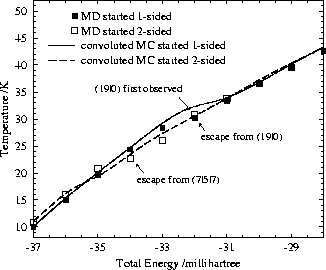
Our MC and the MD results are compared in Fig. 17; they match quite well despite the fact that the results pertain to different ensembles. It is interesting that escape from particular regions of configuration space, a symptom of non-ergodicity, happens at rather similar points in MC and MD except for the sampling of the one-sided regime in initially two-sided standard Monte Carlo simulations. The latter inconsistency probably arises because the number of MC steps was too small to sample all regions of configuration space accessible at this temperature. In simulations at 37.5K with 20 million MC steps we did indeed observe some configurations belonging to the one-sided regime.
The coincidence of the other kinks gives us some confidence that the escape times found in the MD simulations could be experimentally significant. If both MD and MC simulations fail to find structural interconversions it seems unlikely that alternative escape-routes exist. Especially for the one-sided/two-sided transition the fact that MD trajectories starting at very different points produce the same estimate of the escape temperature is probably significant.