For BzAr ![]() , as for the smaller clusters we study the effect of changing the LJ parameters on the
underlying PES.
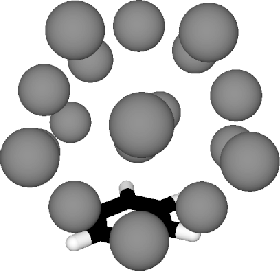
The lowest minimum found, type (7|5|7) (Fig. 3),
is the same two-sided structure for both sets of LJ parameters.
The Ar atoms do not completely surround the benzene molecule in this minimum, and
there is partial `solvation'. We note that the (l|m|n) notation is not able to
describe the geometries for BzAr
, as for the smaller clusters we study the effect of changing the LJ parameters on the
underlying PES.
The lowest minimum found, type (7|5|7) (Fig. 3),
is the same two-sided structure for both sets of LJ parameters.
The Ar atoms do not completely surround the benzene molecule in this minimum, and
there is partial `solvation'. We note that the (l|m|n) notation is not able to
describe the geometries for BzAr ![]() as well as for the smaller clusters,
but it can still be used to differentiate between one-
and two-sided structures or structures involving bridging atoms. Also, unlike the
descriptions of the smaller clusters, this notation no longer implies the existence
of pseudo-planar arrangements of Ar atoms.
Our lowest minimum has some residual double icosahedral structure for the Ar atoms,
but much more icosahedral order is preserved in some higher energy minima
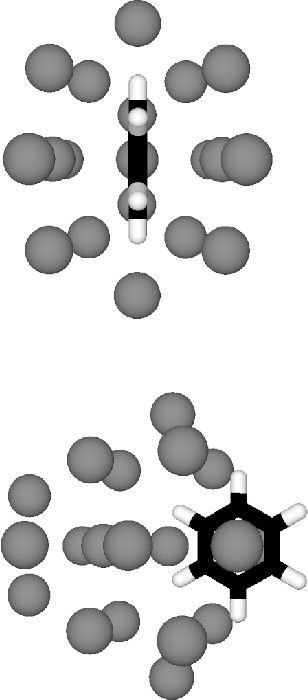
(Fig 4). The double
icosahedron is the global minimum for Ar
as well as for the smaller clusters,
but it can still be used to differentiate between one-
and two-sided structures or structures involving bridging atoms. Also, unlike the
descriptions of the smaller clusters, this notation no longer implies the existence
of pseudo-planar arrangements of Ar atoms.
Our lowest minimum has some residual double icosahedral structure for the Ar atoms,
but much more icosahedral order is preserved in some higher energy minima
(Fig 4). The double
icosahedron is the global minimum for Ar ![]() and when we add a benzene
molecule the effect on the Ar atoms is reduced compared to smaller Ar clusters.
Reordering is localised to Ar atoms in the vicinity of the benzene ring.
and when we add a benzene
molecule the effect on the Ar atoms is reduced compared to smaller Ar clusters.
Reordering is localised to Ar atoms in the vicinity of the benzene ring.
Even though we find the same lowest energy minimum (aside from detailed bond
lengths etc.) for both sets of LJ parameters there are important differences in
the corresponding PES's.
For the OBJ parameters two-sided structures dominate the lowest-lying minima.
However, for the SCM parameters, although the lowest minimum is two-sided, there
exists an energy range of about 0.4millihartree (mh) above this structure
in which we find only one-sided minima.
Such differences in the PES's are likely to
be of some importance when considering the dynamics of the system. We decided to
use the SCM parameters for our Monte Carlo and Molecular Dynamics simulations as we prefer
the larger Ar-Ar well depth and suspect that the smaller Ar-H well depth is more
realistic. The experimental dissocation energy for BzAr ![]() is 1.55mh [44]
which is closer to our SCM value of 1.72mh than our OBJ value of 1.92mh.
is 1.55mh [44]
which is closer to our SCM value of 1.72mh than our OBJ value of 1.92mh.
We start separate simulations from both the lowest-lying two-sided and one-sided minima located by systematic geometry optimisations from preliminary Monte Carlo simulations. These structures are shown in Fig. 3 and Fig. 4. Their binding energies are -39.04mh and -38.90mh respectively. Experimentally there appears to be no definitive conclusion regarding how these clusters form. There may be only one-sided structures initially, which then undergo solvation; alternatively both one-sided and two-sided structures may be formed so that both are observed spectroscopically. In fact, Adams and Stratt [8] have recently suggested that most BzArn clusters formed in a jet are trapped in local minima in which the benzene molecule is bound to the surface of the Ar cluster. This argument is based upon new calculations of the benzene spectral shift which include the collective dielectric response of the cluster. [8] The suggestion that most benzene molecules are trapped in surface states contradicts the original conclusions of Guillaume et al. [3] which, however, have been revised in more recent work (Guillaume et al. 1995).