Next: BzAr Up: LJ parameters Previous: LJ parameters
![]()
![]()
![]()
Next: BzAr
Up: LJ
parameters Previous: LJ
parameters
We systematically search the PES's of BzAr with ![]() using two sets of rather different LJ parameters recommended by Ondrechen,
Berkovitch-Yellin and Jortner [6]
(OBJ) and Schmidt, Le Calvé and Mons [1]
(SCM). In particular the SCM Ar-Ar well depth is about 20% larger than
that used by OBJ and the Ar-H well is about 25% smaller. The equilibrium
separations
using two sets of rather different LJ parameters recommended by Ondrechen,
Berkovitch-Yellin and Jortner [6]
(OBJ) and Schmidt, Le Calvé and Mons [1]
(SCM). In particular the SCM Ar-Ar well depth is about 20% larger than
that used by OBJ and the Ar-H well is about 25% smaller. The equilibrium
separations ![]() are identical for Ar-C and Ar-H and differ by about 3% for Ar-Ar, i.e. these
differences are small compared with the different well depths. These parameters
are summarized in Table i.
are identical for Ar-C and Ar-H and differ by about 3% for Ar-Ar, i.e. these
differences are small compared with the different well depths. These parameters
are summarized in Table i.
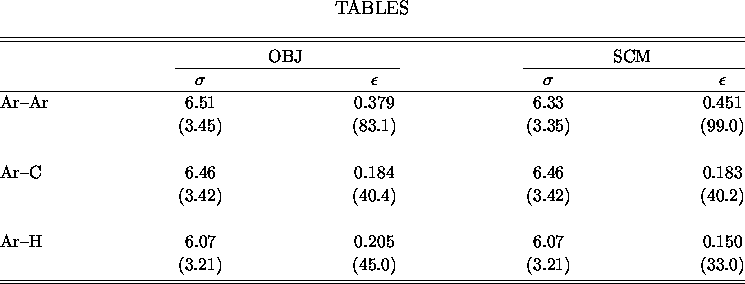
Table: Lennard-Jones parameters from the OBJ [6]
and SCM [1] sources. ![]() is given in bohr(Å) and
is given in bohr(Å) and ![]() is given in millihartree(cm
is given in millihartree(cm ![]() ).
).
We now introduce some notation which has been used, e.g. by Schmidt
et al., to describe the positions of the Ar atoms with respect to the benzene
molecule. Here (l|m|n) signifies that there are l
Ar atoms on one side of the ring, n Ar atoms on the other side and
m Ar atoms in or very close to the plane of the molecule -- the
m Ar atoms are referred to as bridging atoms, and we observe such
structures as local minima for BzAr with ![]() . We note that for structures with no bridging Ar atoms (i.e. m=0)
the notation may be abbreviated to (l|n), where l
and n are defined as above. For example a structure with three Ar
atoms on either side of the benzene molecule is labeled (3|3) and if an
additional Ar atom is placed in the plane of the molecule this is referred
to as a (3|1|3) structure.
. We note that for structures with no bridging Ar atoms (i.e. m=0)
the notation may be abbreviated to (l|n), where l
and n are defined as above. For example a structure with three Ar
atoms on either side of the benzene molecule is labeled (3|3) and if an
additional Ar atom is placed in the plane of the molecule this is referred
to as a (3|1|3) structure.
Following Schmidt et al. we use two additional labels c
and s for describing the arrangement of the Ar atoms with respect
to the ![]() axis of the benzene molecule. We use c when one of the Ar atoms
lies on or close to this axis and s when this point is `shared'
by more than one Ar atom. For c-type arrangements of Ar atoms those
not above the
axis of the benzene molecule. We use c when one of the Ar atoms
lies on or close to this axis and s when this point is `shared'
by more than one Ar atom. For c-type arrangements of Ar atoms those
not above the ![]() axis are positioned with the benzene molecule acting as a template -- each
atom lies in or near to one of the mirror planes between the H atoms. This
leads to closed-packed arrangements of Ar atoms, which we would expect
to see up to BzAr
axis are positioned with the benzene molecule acting as a template -- each
atom lies in or near to one of the mirror planes between the H atoms. This
leads to closed-packed arrangements of Ar atoms, which we would expect
to see up to BzAr ![]() for one-sided structures, at which point the benzene ring becomes `saturated'.
We observe s-type minima for BzAr
for one-sided structures, at which point the benzene ring becomes `saturated'.
We observe s-type minima for BzAr ![]() to BzAr
to BzAr ![]() and for the transition state
and for the transition state ![]() in BzAr
in BzAr ![]() , which connects two permutational isomers of the
, which connects two permutational isomers of the ![]() local minimum in a degenerate rearrangement in agreement with previous
calculations by Wales [2].
local minimum in a degenerate rearrangement in agreement with previous
calculations by Wales [2].
The additional labels s and c are useful to describe many
of the minima that we observe in the smaller BzAr clusters because of the
influence that the benzene molecule has on the arrangement of the Ar atoms.
For two-sided structures it is generally the case that the arrangement
of Ar atoms on each side of the molecule is similar to one observed for
a one-sided local minimum in a smaller cluster. For example, local minima
exist for ![]() and
and ![]() structures and as indicated in the discussion above we also observe
structures and as indicated in the discussion above we also observe ![]() minima. This notation does not indicate the relative orientations of the
two sets of Ar atoms and hence several local minima generally exist with
the same label in such cases.
minima. This notation does not indicate the relative orientations of the
two sets of Ar atoms and hence several local minima generally exist with
the same label in such cases.
The majority of the low-lying local minima for the smaller clusters
have pseudo-planar arrangements of Ar atoms on each side of the ring, i.e. each
atom is approximately the same perpendicular distance from the plane of
the benzene molecule. Some structures have a second plane containing one
or two Ar atoms above the first. An example for which three Ar atoms are
in an s-type arrangement with the fourth acting as a cap would be
labelled ![]() .
.
This nomenclature allows us to describe many of the local minima that
we observe for up to eight Ar atoms. However, for clusters as small as
BzAr ![]() we observe structures not based primarily on the geometry of the benzene
molecule and there are local minima with pseudo 5-fold symmetric arrangements
of Ar atoms. For BzAr
we observe structures not based primarily on the geometry of the benzene
molecule and there are local minima with pseudo 5-fold symmetric arrangements
of Ar atoms. For BzAr ![]() we see structures based on the neat Ar
we see structures based on the neat Ar ![]() double icosahedron as discussed below.
double icosahedron as discussed below.
Details of some low-lying minima, along with their point groups and
energies, are given in Table ii.
Details of sample reaction pathways are given in Table iii.
It is noteworthy that one-sided minima are found for which the expected
close-packed arrangement of Ar atoms does not exist, i.e. minima for
which the Ar atoms are spread above the benzene molecule in patterns which
do not favour the Ar-Ar interactions. We find such structures for BzAr
with ![]() and these, as for the s- and c-type arrangements, are able
to combine with other Ar atoms to form two-sided structures. For example,
a minimum with a linear arrangement of Ar atoms above the benzene molecule
exists. We label this
and these, as for the s- and c-type arrangements, are able
to combine with other Ar atoms to form two-sided structures. For example,
a minimum with a linear arrangement of Ar atoms above the benzene molecule
exists. We label this ![]() structure
structure ![]() and have also observed
and have also observed ![]() . The unfavourable arrangements of Ar atoms in these minima make them relatively
weakly bound.
. The unfavourable arrangements of Ar atoms in these minima make them relatively
weakly bound.
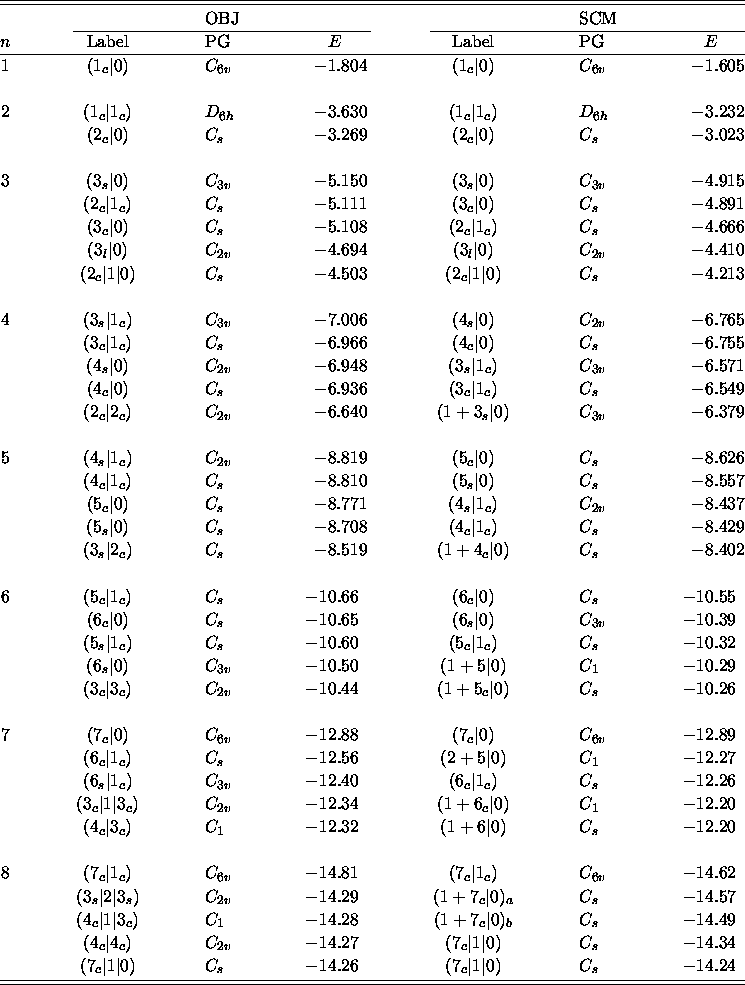
Table ii: The energies, in millihartree, and the point groups (PG)
for the five lowest BzAr minima found with the OBJ and SCM LJ parameters.
The labels are fully described within the text.
The geometries of the minima and the transition states, calculated without
the induction energy contribution, are qualitatively unaffected in almost
all cases when we change the LJ parameters from those of OBJ to SCM. Occasionally
a structure characterized for one set of LJ parameters as a minimum is
not so for the other set and sometimes the connectivity of the minima changes,
i.e. which minima are linked by particular transition states. Although
the structures generally do not change very much, we find that the energetic
ordering of the local minima is rather different, and we observe that the
SCM minima show a preference for one-sided structures compared to those
of OBJ. The SCM lowest minima for BzAr with ![]() are all one-sided, with close-packed, pseudo-planar arrangements of Ar
atoms. The size of the Ar-Ar well depth accounts for the fact that this
trend was not observed for the OBJ lowest energy minima where two-sided
structures have greater stability. For example, the
are all one-sided, with close-packed, pseudo-planar arrangements of Ar
atoms. The size of the Ar-Ar well depth accounts for the fact that this
trend was not observed for the OBJ lowest energy minima where two-sided
structures have greater stability. For example, the ![]() structure is lowest in energy for BzAr
structure is lowest in energy for BzAr ![]() for both SCM and OBJ but for BzAr
for both SCM and OBJ but for BzAr ![]() and BzAr
and BzAr ![]() the lowest minima found with the SCM parameters are
the lowest minima found with the SCM parameters are ![]() and
and ![]() and with the OBJ parameters are
and with the OBJ parameters are ![]() and
and ![]() -- see Table ii.
-- see Table ii.
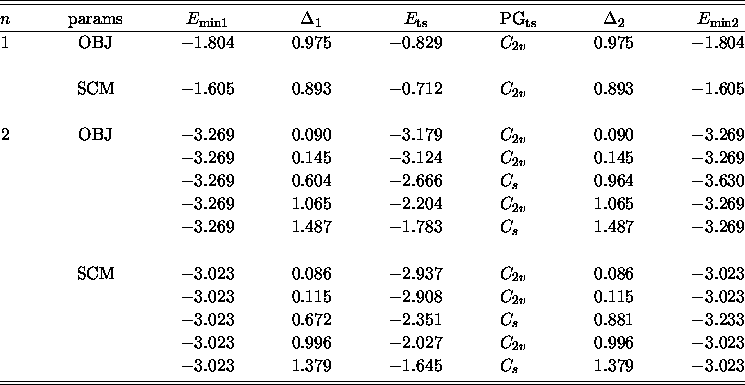
Table iii: Selected rearrangement mechanisms for some small BzAr
clusters. We report the energies of the minima ( ![]() and
and ![]() ), the energy of the transition state (
), the energy of the transition state ( ![]() ), the barrier heights (
), the barrier heights ( ![]() and
and ![]() ), and the transition state point groups (PG
), and the transition state point groups (PG ![]() ). All energies in millihartree.
). All energies in millihartree.
For BzAr with ![]() we see differences in the transition states calculated using the different
parameters. For BzAr
we see differences in the transition states calculated using the different
parameters. For BzAr ![]() we find five transition states for both OBJ and SCM. Four are degenerate
rearrangements of the
we find five transition states for both OBJ and SCM. Four are degenerate
rearrangements of the ![]() minimum and the other is non-degenerate, exchanging the
minimum and the other is non-degenerate, exchanging the ![]() and the
and the ![]() minima. There is a correlation between the energies of the transition states
and the number of Ar atoms crossing the benzene ring. The two highest-lying
transition states involve both Ar atoms crossing the ring and mediate degenerate
rearrangements. The next highest transition state is for the non-degenerate
rearrangement in which one Ar atom changes sides. The remaining two degenerate
rearrangements involve no ring-crossing and in the higher of the two the
benzene molecule effectively rotates by
minima. There is a correlation between the energies of the transition states
and the number of Ar atoms crossing the benzene ring. The two highest-lying
transition states involve both Ar atoms crossing the ring and mediate degenerate
rearrangements. The next highest transition state is for the non-degenerate
rearrangement in which one Ar atom changes sides. The remaining two degenerate
rearrangements involve no ring-crossing and in the higher of the two the
benzene molecule effectively rotates by ![]() , thus changing the site of one of the Ar atoms. The lower of the latter
two transition states corresponds to a
, thus changing the site of one of the Ar atoms. The lower of the latter
two transition states corresponds to a ![]() structure and the corresponding rearrangement changes the Ar atom lying
on the
structure and the corresponding rearrangement changes the Ar atom lying
on the ![]() axis of the molecule. The transition states permute the Ar atoms in the
same way for both the OBJ and SCM parameters, and with one exception the
rearrangements are equivalent. During the
axis of the molecule. The transition states permute the Ar atoms in the
same way for both the OBJ and SCM parameters, and with one exception the
rearrangements are equivalent. During the ![]() rotation, for the SCM parameters one Ar atom remains on the
rotation, for the SCM parameters one Ar atom remains on the ![]() axis of the benzene molecule. However, for the OBJ parameters the rotation
of the benzene molecule occurs after the
axis of the benzene molecule. However, for the OBJ parameters the rotation
of the benzene molecule occurs after the ![]() structure has been reached and therefore involves a direct connection between
two transtion states. If we label the
structure has been reached and therefore involves a direct connection between
two transtion states. If we label the ![]() minimum min, the
minimum min, the ![]() transition state ts1 and the second transition state ts2,
then we can label the two low-lying OBJ pathways as,
transition state ts1 and the second transition state ts2,
then we can label the two low-lying OBJ pathways as,
though the right hand side minimum is not the same permutational isomer for the two paths (Fig. 2). Branching points must exist between ts2 and ts1 on both sides of the second path. We find such rearrangements involving two transition states for the OBJ parameters in BzAr with n=2-3,5.
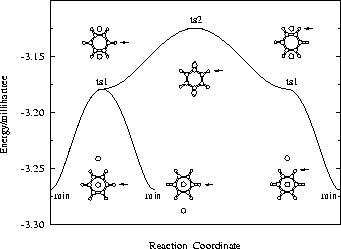
Figure 2: Two rearrangement pathways for BzAr ![]() calculated using the OBJ parameters. One includes a direct connection between
two transition states. Both pathways are for degenerate rearrangements
of the
calculated using the OBJ parameters. One includes a direct connection between
two transition states. Both pathways are for degenerate rearrangements
of the ![]() minimum. The arrows point to the same H atom in all cases, to clarify the
relative orientations of the benzene molecule.
minimum. The arrows point to the same H atom in all cases, to clarify the
relative orientations of the benzene molecule.
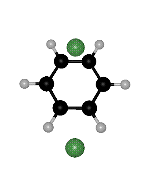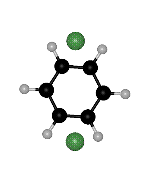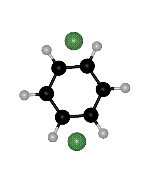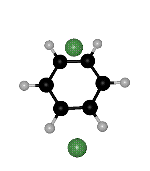 Click on the picture for animation: min -> ts1 -> ts2 -> ts1 ->
min
Click on the picture for animation: min -> ts1 -> ts2 -> ts1 ->
min
We will cite one other difference in the PES's. For BzAr ![]() there exists a transition state which converges to a different minimum
in one of the two downhill optimizations. For the OBJ parameters, the minima
on either side of this transition state are
there exists a transition state which converges to a different minimum
in one of the two downhill optimizations. For the OBJ parameters, the minima
on either side of this transition state are ![]() and
and ![]() . For the same transition state using the SCM parameters, the minima it
connects are
. For the same transition state using the SCM parameters, the minima it
connects are ![]() and
and ![]() . If we follow the reaction pathway for these parameters we find that the
. If we follow the reaction pathway for these parameters we find that the
![]() structure is visited but it is not a stationary point and the optimization
continues until the
structure is visited but it is not a stationary point and the optimization
continues until the ![]() structure is reached.
structure is reached.
In conclusion we find that as well as the variation in the energetic ordering of the stationary points, examples for which the OBJ and SCM PES's have other fundamental differences exist. However, these are rare, and it appears that even considerable changes in the LJ parameters leave the form of the stationary points largely unaffected. For brevity we have only tabulated a small fraction of the results we have obtained for the various stationary points.