Recent advances in simulation techniques have made it possible to begin to
study dense polymer systems. In particular, we use configurational-bias
Monte Carlo[25] including moves in which a mid-section
of the chain is regrown.[26]
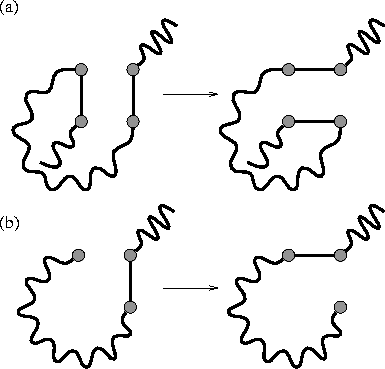
We also make occasional bond-flipping moves [Figure 1] which,
although they do not change the shape of the volume occupied by the polymer,
change the path of the polymer through that volume.[27,28]
These moves speed up equilibration in the dense phases.
The simulation method was tested by comparing to results obtained for ![]() by exact enumeration.[29,30]
Thermodynamic properties, such as the heat capacity, were calculated from the energy
distributions of each run using the multi-histogram method.[31,32]
by exact enumeration.[29,30]
Thermodynamic properties, such as the heat capacity, were calculated from the energy
distributions of each run using the multi-histogram method.[31,32]
 |
The number of Monte Carlo steps used in our simulations
was typically 4-30![]() .
The longer simulations were required for the larger polymers,
especially at temperatures where two or more states coexisted.
The sizes of the polymers we studied were N=27, 64, 100, 216, 343.
These sizes are `magic numbers' for
.
The longer simulations were required for the larger polymers,
especially at temperatures where two or more states coexisted.
The sizes of the polymers we studied were N=27, 64, 100, 216, 343.
These sizes are `magic numbers' for ![]() , because
compact cuboids of low aspect ratios can be formed at these sizes.
In the presentation of our results we concentrate on polymers with N=100 and 343,
the former because the smaller size allows for clear visualization of the structures
of the different phases, and the latter because the effects of finite size will
be smallest.
, because
compact cuboids of low aspect ratios can be formed at these sizes.
In the presentation of our results we concentrate on polymers with N=100 and 343,
the former because the smaller size allows for clear visualization of the structures
of the different phases, and the latter because the effects of finite size will
be smallest.
In order to monitor the orientational order within the polymer we devised an order parameter, Q, which is given by
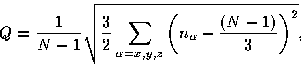 |
(6) |