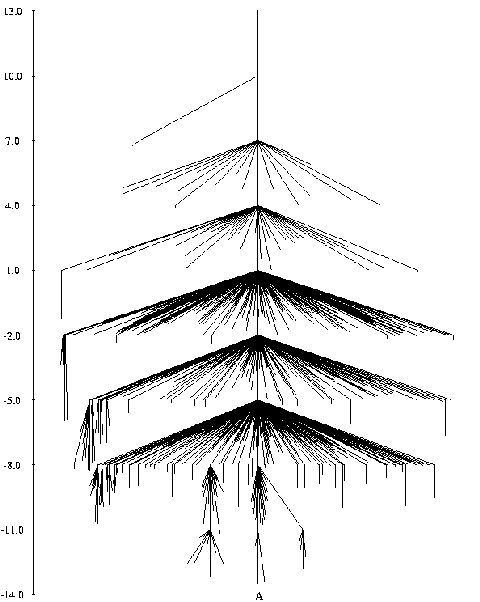
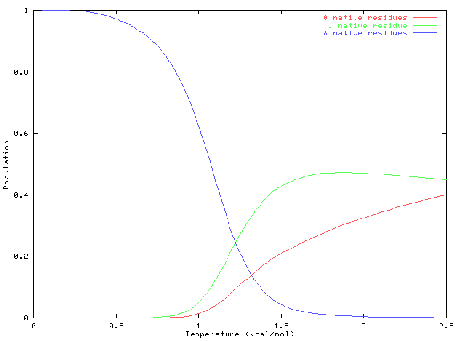
It is widely believed that the native conformation of a protein is the conformation with the lowest free energy, i.e. the global minimum of the free energy surface. This is known as Anfinsen's thermodynamic hypothesis. It is rather difficult (and expensive) to calculate free energies - by definition these involve averaging over a large number of conformations - so we instead concentrate on potential energies, making the assumption that the global free energy minimum should be a very low energy minimum on the PES, if not always the global minimum. Potential energies are functions only of atomic coordinates and are thus much easier to calculate. Efficient algorithms for global optimization of the PES should therefore be very useful tools for protein structure prediction.
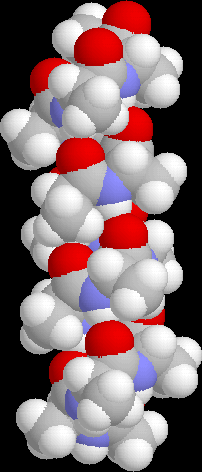
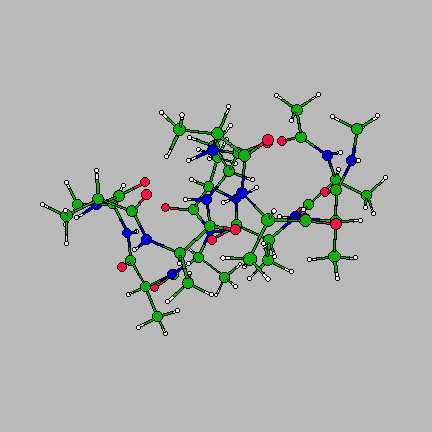
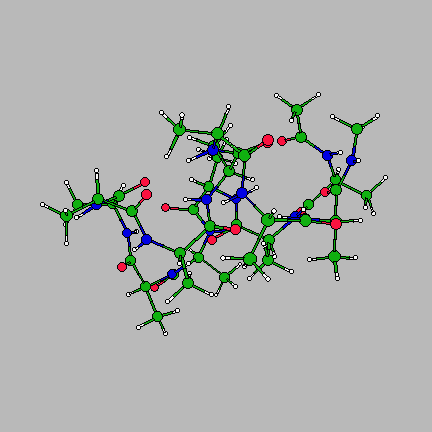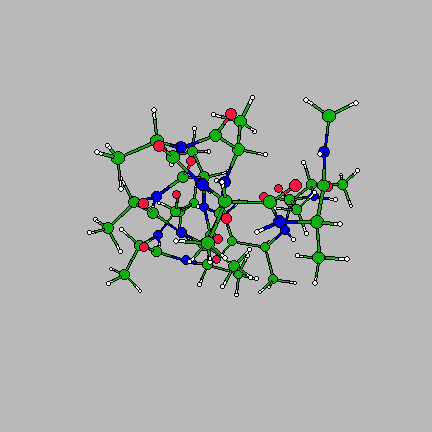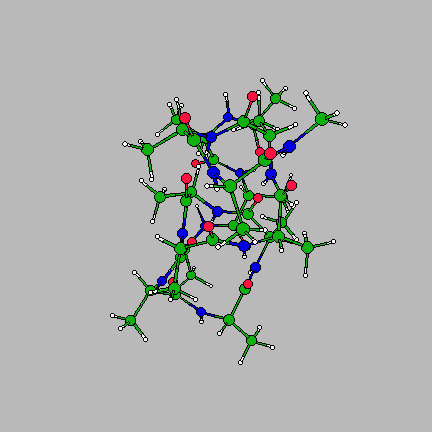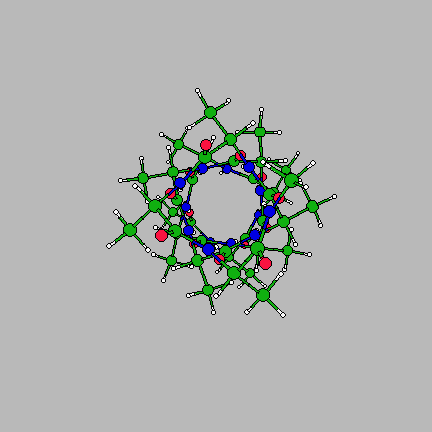
The image on the right is of the global minimum for Ac-(ala)14-NHMe using the AMBER95 force field with a dielctric constant of 1. This was located using the basin-hopping algorithm of Wales and Doye (incorporated in the program GMIN), which is essentially the Monte-Carlo with Minimization algorithm of Scheraga and Li. Recently I have been working on a parallel "taboo" algorithm, where several basin-hopping runs are set in motion at once. If any run gets too close in conformation space to another run, its coordinates are randomly reseeded. The algorithm thus attempts to maximise the volume of conformation space which is explored, while at the same time allowing each individual run to explore its area of conformation space thoroughly.
Click here for a list of (what I believe to be) global minima for some poly-(L)-alanine chains and their structures.
{kind=link}
{kind=link}