- ADDMIN file: reads in the data for one or more minima from file
in OPTIM min.data.info format and then stops.
Any number of min.data.info files can be concatenated into file.
New entries will be created in min.data and points.min.
If min.A and min.B do not exist they are not created.
READMIN therefore provides an alternative way to start an initial
path calculation by providing minima, so long as min.A and min.B
are created by hand.
It also provides a way to add additional minima to an existing database, since
new entries should be appended. There is a check to make sure that the minima
added are different from all those currently known.
This keyword has the same function as READMIN and is provided for convenience.
- ADDMINXYZ file: reads coordinates in xyz format from
file file and runs OPTIM for each one using odata.addminxyz,
which needs to contain the OPTIM keywords DUMPDATA and ENDHESS
so that a min.data.info file is created.
The resulting min.data.info files are then used to add minima to the
database, which can be empty initially. CYCLES must also be set to a non-zero value.
- ADDPATH afile : reads pathway data from file afile.
If ADDPATH is used on its own before a connection has been
achieved then DIJINITCONT should be specified so that the
pairdist and pairlist files are written.
- ADDPERM: adds permutational isomers of every
stationary point to the databases. Not recommended except for small systems
where you are using a tagged atom to study an isomerisation (e.g. 2D LJ7).
- ADDPERM2 start finish: adds all distinct permutation-inversion isomers of every
stationary point to the databases for permutations of atoms numbered from
start to finish, inclusive. Not recommended except for small systems!
Added this capability to allow studies where permutation-inversion isomers are
not automatically lumped togther, to see the effect of free energy regrouping
(or observation time scale) on the landscape.
Any perm.allow file present is ignored.
- ADDPERM3: adds all distinct permutation-inversion isomers of every
stationary point to the databases according to the allowed permutations in
perm.allow.
Can be used in combination with MERGEDB to create a new database where
permutation-isomers are distinguised.
- ALLOWAB: allows minima to belong to both A and B sets where lumping
is performed with REGROUPFREE and RFMULTI.
This may be useful for understanding how free energy groups merge as a function of
observation time scale and temperature, when rate constants are not required.
- ALLTS : specifies that transition states that are considered identical by energy
and geometry conditions to one already in the database, but which connect different minima are not discarded
and are treated as a new transition state. This is especially useful when starting PATHSAMPLE from an
OPTIM path.info file using STARTFROMPATH and STARTTRIPLES, as the path.info file from
OPTIM may contain such transition states, even if only one is involved in the connected path.
- ANGLEAXIS : specifies angle-axis coordinates for rigid bodies. NB: this is not part of the GENRIGID framework.
- AMBER9 : specifies the AMBER 9 potential.
- AMH : specifies an AMH potential.
- AMHQ whichmin : Calculates Wolynes Q score based on the native state structure.
- AMHRMSD whichmin : Calculates RMSD based on the native state structure.
- AMHQCONT whichmin rcut : Calculates Q-cutoff score based on a native distance.
- AMHRELQ whichmin1 whichmin2 : Calculates Wolynes Q score between two minima.
- AMHALLATOM : Adds sidechains and completes backbone for AMH structures with SCWRL.
Input structure is amhmin.pdb. Output structure is amhmin.pdb.scwrl. Scwrl output logs scwrl_out.
- AMH_RELCO whichmin rcut : Calculates relative contact order for AMH structures.
- ATOMMATCHDIST: Currently for bulk systems only, providing an alternative method for finding the shortest distance between structures.
This method is particularly useful for finding the shortest distance between very similar structures such as defective crystal structures.
The method is an extension of the original bulkmindist.f90 routine for matching permutational isomers. Atoms are overlayed and the number of exactly matching atoms is maximised.
With ATOMMATCHDIST, the method is non-deterministic to maximise efficiency. It has been optimised to work particularly well for providing small distances
between similar structures and for removing very large distances. However, it will not always outperform the default method for middling distances.
Currently only one method for calculating distances, atom matching or the default, can be used.
- ATOMMATCHFULL: As for ATOMMATCHDIST, but provides a slow deterministic result that should always outperform or equal the default distance calculation and ATOMMATCHDIST but may never finish.
- BHINTERP dthresh maxe bhsteps conv T stepsize accrat K sfrac ICINTERP: specifies that
OPTIM jobs should be run using only the BHINTERP interpolation option,
where no transition state searches are run. Requires the auxiliary file odata.bhinterp.
This option is intended for use with DIJINITSTART and DIJINITCONT.
The various arguments are used to add a BHINTERP line to the OPTIM odata file.
The distance threshold parameter specifies that interpolation between consecutive minima
in the Dijkstra list should occur if their minimum distance is greater than dthresh.
The threshold actually used in the corresponding odata file is either the minimum distance
divided by two or dthresh, whichever is greater. This arrangement enables
parallel OPTIM jobs to be run for different pairs of minima.
Intermediate minima are only accepted if their energy is below maxe.
A basin-hopping global optimisation run of bhsteps is run for each pair of
end point minima within dthresh using an RMS gradient convergence criterion
of conv, a temperature parameter of T, and a maximum step size for
perturbations of stepsize. The step size is adjusted dynamically towards an
acceptance ratio target of accrat.
The objective function consists of the energy of the minimum on the potential energy surface
plus the energy corresponding to harmonic springs of force constant K
stretched to displacements corresponding to the minimum distance between the new minimum
and the end points.
sfrac is used in the initial interpolation: a value of 0.5 will put the initial
guess half-way between the end points, and in general the geometry will be sfrac times one
end point plus
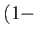 sfrac
sfrac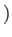 times the other.
For large distances, using a value other than a half may be helpful.
If ICINTERP is present then amino acid side chains are interpolated using
internal coordinates for CHARMM runs.
The step size is interpreted in degrees for CHARMM, where the perturbations used
to step between minima are performed using dihedral angle twists.
The algorithm is applied recursively between minima as new minima are found.
A new minimum will not be accepted if both distances to the minima we are
currently trying to interpolate between are greater than the minimum distance
between these minima.
times the other.
For large distances, using a value other than a half may be helpful.
If ICINTERP is present then amino acid side chains are interpolated using
internal coordinates for CHARMM runs.
The step size is interpreted in degrees for CHARMM, where the perturbations used
to step between minima are performed using dihedral angle twists.
The algorithm is applied recursively between minima as new minima are found.
A new minimum will not be accepted if both distances to the minima we are
currently trying to interpolate between are greater than the minimum distance
between these minima.
- BISECT dthresh maxe bisectsteps attempts ICINTERP: specifies that
OPTIM jobs should be run using only the BISECT interpolation option,
where no transition state searches are run. Requires the auxiliary file odata.bisect.
This option is intended for use with DUMMYTS.
The various arguments are used to add a BISECT line to the OPTIM odata file.
The distance threshold parameter specifies that bisection between consecutive minima
is attempted if their minimum distance is greater than dthresh.
The interpolated geometry is minimised and compared with the starting structures.
New minima are only accepted if their energy is below maxe; they are added to the
current list of known minima in the OPTIM job between the two starting structures.
The estimated energy of any dummy transition states between the two starting structures
is raised if the sum of distances between the new minimum and the two structures is
greater than the original minimised distance.
bisectsteps is the maximum number of bisection steps, and
attempts is the number of attempts per step for a given pair.
If minimisation leads to one of the end points the interpolation is changed to use
fractions of the two structures that become increasingly skewed, as for
the adjustment of sfrac with BHINTERP, above.
The ts.data file is rewritten if the any dummy transition state energies are revised.
If ICINTERP is present then amino acid side chains are interpolated using
internal coordinates for CHARMM runs.
- BULK x1 x2 x3: a bulk system in a periodic box of lengths x1, x2, x3.
- CALCORDER: the program will call subroutine calcorder.f90 and calculate
order parameters, before exiting. Since the order parameters will be system dependent
users must compile in an appropriate calcorder.f90 themselves.
- CAPSID rho eps rad h: parameters for a rigid pentagonal pyramid. If
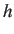 is omitted
it is set to half
is omitted
it is set to half 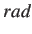 by default.
by default.
- CHARMM: specifies that the CHARMM potential is used in the OPTIM runs.
- CHECKCONECTIONS: instruction to check that each minimum has at least the number
of connections specified by the CONNECTIONS keyword at the start of every run,
before anything else is done. See CONNECTIONS below.
This keyword results in calls to the original single-ended transition state
searching subroutine, which is not efficiently parallelised.
The NEWCONNECTIONS keyword should probably be used instead.
- CHECKMIN start finish: will rerun OPTIM for all the
minima from start to finish using the odata file
odata.checksp, which must be present in the working directory.
PATHSAMPLE will report whether or not the OPTIM job converged, so the number
of minimization steps should be set accordingly (e.g. BFGSSTEPS 0).
This option may be useful for finding any unconverged minima in a corrupted database,
which could then be removed using REMOVESP.
- CHECKTS start finish: will rerun OPTIM for all the
transition states from start to finish using the odata file
odata.checksp, which must be present in the working directory.
PATHSAMPLE will report whether or not the OPTIM job converged.
odata.checksp should probably be the same as for CHECKMIN, as in, a minimization
with zero steps where only the initial energy and gradient are calculated to test for convergence
and the Hessian index is NOT checked.
This option may be useful for finding any unconverged transition states in a corrupted database,
which could then be removed using REMOVESP.
- CLOSEFILES: open and close the min.data and ts.data files as needed.
By default these files stay open all the time. For small systems this inevitably seems to cause
file corruption on nfs mounted file systems, which is apparently due to bugs in the interaction
of nfs with the Linux kernel. The symptom is that strings of control characters appear randomly in
min.data and/or ts.data, which means that the run cannot continue without
manually fixing the file in question.
The CLOSEFILES keyword has provided a successful workaround for this problem.
- CONNECTIONS n maxattempts: minimum number of connections for each minimum.
For each new minimum found PATHSAMPLE will attempt to find n connected minima.
If n is one or fewer then no additional single-ended transition state searches are
needed, and the more efficient cycle2.f90 subroutine is used. In this case, OPTIM
jobs do not have to be processed in batches; instead, a new OPTIM job is submitted
as soon as one finishes. The default for n is now 0.
The maximum number of transition state searches from each new minimum is maxattempts,
for which the default is 10.
Note that you will need file odata.tssearch in the current
working directory if n is greater than one.
The odata.tssearch file needs to contain OPTIM PATH
and DUMPALLPATHS directives
so that a path.info file is created.
It is no longer necessary to supply a separate odata.path file.
This keyword results in calls to the original single-ended transition state
searching subroutine, which is not efficiently parallelised.
The NEWCONNECTIONS keyword should probably be used instead.
- CONINT specifies that terms for internal minimum distances
should be included in the energy and gradient for the auxiliary interpolation
potential when INTCONSTRAINT is specified.
- CONNECTREGION min1 min2 dist: grows the stationary point database by running
double-ended transition state searches between minima whose minimum separation is less then
dist. min1 and min2 specify the starting minima in the double loop over
pairs, so that restarted runs need not consider pairs that have previously been searched.
Minima that already have a direct connection are ignored.
The maximum number of candidate pairs calculated per call to connectd.f90 is 10000.
This parameter may be useful for exhaustive searches for small systems.
- COPYFILES a1 a2 a3
 : specify additional files that need to be
copied to distributed nodes. Only files odata.<pid> and finish.<pid> are
copied by default. For example, the BLN potential requires file BLN sequence, while
CHARMM runs may need one or more crd files. Each string must be less than or equal to
twenty characters in length, and the total, including separating blanks, must not exceed
80 characters. If the CHARMM keyword is set and the COPYFILES
arguments do not include input.crd then the program will stop.
: specify additional files that need to be
copied to distributed nodes. Only files odata.<pid> and finish.<pid> are
copied by default. For example, the BLN potential requires file BLN sequence, while
CHARMM runs may need one or more crd files. Each string must be less than or equal to
twenty characters in length, and the total, including separating blanks, must not exceed
80 characters. If the CHARMM keyword is set and the COPYFILES
arguments do not include input.crd then the program will stop.
- COPYOPTIM: if present, some of the OPTIM output files will be copied to
the mother superior node and not deleted. This behaviour also occurs if the DEBUG
keyword is present. The copied files, in addition to path.info (min.data.info
for a BHINTERP run), are the OPTIM output file, the odata file
and the finish file.
- CPUS ncpu: specifies the number of cpu's (cores) available when running
PATHSAMPLE interactively, without using a scheduler such as PBS to
send jobs to distributed memory nodes. If used on
a distributed memory cluster then n OPTIM jobs will be run at any given
time, and they will all run on the interactive node in question.
Can be used to run multiple OPTIM jobs on an interactive testing node of a distributed memory
machine.
- CYCLES n: the number of cycles of ncpu or
jpn
 nodes OPTIM jobs to run in sampling the stationary point
database.
nodes OPTIM jobs to run in sampling the stationary point
database.
- CV Tmin Tmax Tinc: calculate the heat capacity in the harmonic
superposition approximation from temperature Tmin to Tmax at
intervals of Tinc.
- DB sigmabb: calls
a finite system of dipolar Lennard-Jones dumbbells [11], where
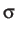 's correspond to the Lennard-Jones parameters.
's correspond to the Lennard-Jones parameters. 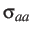 is set to unity by default.
is set to unity by default.
- DEBUG: turn on debug printing.
- DEGFREEDOMS n: used to specify the number of degrees of freedom, n, in the system. This
should not be used with RIGIDINIT. Instead, the number of degrees of freedom should be specified as an argument
to that keyword.
- DGT DisconnectSources AltPbb Rescale Normalise:
dense optimised graph transformation rate calculation [4].
The four arguments are all logicals, so an example input line might look
DGT T T F F. If true (the default) DisconnectSources specifies that
a full transformation should be performed for each source, disconnecting the
other sources. The resulting rate constants correspond to the KMC result and
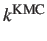 [5].
If AltPbb is true (default is false) then additional work is done to
maintain precision, which roughly doubles the execution time, but may be needed at low
temperature.
If Rescale is true (default false) an alternative strategy is used
to try and prevent error propagation.
Setting Normalise true (default false) instructs GT2input.f90 to check the normalisation
of branch probabilities. This should not be necessary.
[5].
If AltPbb is true (default is false) then additional work is done to
maintain precision, which roughly doubles the execution time, but may be needed at low
temperature.
If Rescale is true (default false) an alternative strategy is used
to try and prevent error propagation.
Setting Normalise true (default false) instructs GT2input.f90 to check the normalisation
of branch probabilities. This should not be necessary.
- DIJINITCONT exp or n max p1 p2: continue an initial Dijkstra connection
from information in the existing min.A, min.B, min.data,
ts.data, points.ts and points.min files.
The minimum information required consists of database entries
for the two endpoint minima.
The algorithm used is similar to OPTIM, except that the missing connections can
be sought in parallel OPTIM runs, each of which is itself a separate
Dijkstra connect calculation.
If exp appears after DIJINIT then the edge weights are based on the
exponential distance; otherwise the distance is raised to the power n, which
defaults to 2.
The max parameter (default 10) specifies the number of entries for each
minimum in the sorted list of metric values (default metric is distance).
These values are now dumped in the file pairdist with the identities of the
minima stored in file pairlist.
This scheme enables much faster restarts if the number of minima grows large before
a connection is found, and limits the memory used to linear scaling rather than quadratic.
If ADDPATH is used on its own before a connection has been
achieved then DIJINITCONT should be specified so that the
pairdist and pairlist files are written.
If the same connection is attempted more than once the same odata file
is used, and the previous calculation is simply duplicated.
The number of parallel searches is equal to the number of processors available.
For the first cycle all the searches would be the same, because there must be a single
gap between the two end minima.
To avoid unnecessary duplication it is best to perform an initial run on
one processor, or a single node, for one cycle (see DIJINITSTART below).
Then restart by simply increasing the number of cycles in the pathdata file
as well as the number of processors.
In the Dijkstra analysis minima with zero connections are removed.
This value can be changed using a DIJKSTRA n line in pathdata.
If p1 and p2 are specified then PATHSAMPLE will calculate the
pairlist and pairdist entries for minima in the range p1 to
p2 (inclusive) write them to files pairlist.p1.p2 and pairdist.p1.p2,
and stop.
- DIJINITCONTFLY exp or n: performs the same function as DIJINITCONT but
calculates distances between local minima on the fly. This approach may be necessary
for initial path runs that produce more that 20,000 minima, where fitting all the
distances into memory becomes an issue.
- DIJINITSTART exp or n max: perform an initial Dijkstra connection run
for the two endpoints specified in odata.start and odata.finish.
In this case the files min.A, min.B, min.data, ts.data (and all
points files) will be created automatically.
Any existing database information will be overwritten.
Note that the odata.start and odata.finish files must contain
the OPTIM keyword DUMPDATA and calculate the normal mode frequencies.
For a CHARMM run the files start.crd and finish.crd are also needed.
The algorithm used is similar to OPTIM, except that the missing connections can
be sought in parallel OPTIM runs, each of which is itself a separate
Dijkstra connect calculation.
If exp appears after DIJINIT then the edge weights are based on the
exponential distance; otherwise the distance is raised to the power n, which
defaults to 2.
The max parameter (default 10) specifies the number of entries for each
minimum in the sorted list of metric values (default metric is distance).
These values are now dumped in the file pairdist with the identities of the
minima stored in file pairlist.
If ADDPATH is used on its own before a connection has been
achieved then DIJINITCONT should be specified so that the
pairdist and pairlist files are written.
This scheme enables much faster restarts if the number of minima grows large before
a connection is found, and limits the memory used to linear scaling rather than quadratic.
If the same connection is attempted more than once the same odata file
is used, and the previous calculation is simply duplicated.
The number of parallel searches is equal to the number of processors available.
For the first cycle all the searches will be the same, because there must be a single
gap between the two end minima.
To avoid unnecessary duplication it is usual to perform the first cycle of an initial
connection attempt on one processor for one cycle.
Then restart using DIJINITCONT
by simply increasing the number of cycles in the pathdata file
as well as the number of processors.
For the first cycle it is also a good idea to use just one double-ended connection
cycle in odata.connect, and increase this value on subsequent cycles
when some intervening minima are known.
Otherwise the first OPTIM job may spend a great deal of time performing
connection attempts that could be spread over multiple processors in a
PATHSAMPLE DIJINITCONT job.
In the Dijkstra analysis minima with zero connections are removed.
This value can be changed using a DIJKSTRA n line in pathdata.
- DIJINITSTARTFLY exp or n: performs the same function as DIJINITSTART but
calculates distances between local minima on the fly. This approach may be necessary
for initial path runs that produce more that 20,000 minima, where fitting all the
distances into memory becomes an issue.
- DIJKSTRA n: perform a Dijkstra analysis to find the path
with the largest contribution to the `SS' rate constant ignoring recrossings. Minima with
n connections or fewer are pruned.
If REGROUPFREE, REGROUPRATE, or REGROUPPE is present
in pathdata then the stationary points will be regrouped first. In this
case the energies in Epath are free energies, and files such as
redopoints and stationary.points are not produced.
Once a path is found the rate corresponding to that path in isolation,
i.e. ignoring other connections, is calculated using the graph transformation
method.[4].
- DIJKSTRAWAIT: perform a Dijkstra analysis to find the path
with the lowest sum of waiting times times initial conditional probability.
- DIJPAIR n1 n2 string: choose pairs of minima for connection attempts based on
the highest barrier on the path that makes the largest contribution
to the `SS' rate constant. The first time a given transition state is the highest,
the connection attempt is between the adjacent minimum.
The next time it is chosen we use the second-nearest neighbours along the path in question.
The parameter n1 determines how many steps away from the transition state we
continue these connection attempts for. The default is n1=1.
Once n1 attempts have been tried for the highest transition state we
move to the second-highest, and so on.
Minima with n2 connections or fewer are iteratively removed before the network
analysis. The default value for n2 is one.
The string argument, which is optional, only has an effect if it is the
word `BARRIER'. In this case the pairs of minima to connect are sorted according to the
largest barrier
that separates them. Otherwise they are sorted in order of increasing
energy of the minimum that is separated from the product region.
- DIRECTION a: the direction (`AB' or `BA') determines the sense of a path, for
contexts where forward and backward or reactant and product need to be defined. Note the use
of the spectroscopists' convention, whereby `AB' means from B to A . The default is `AB'.
- DSCALE x: specifies how the separation between local minima, d, is used
to select the pair used in the next attempted connection. If
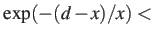 a random number drawn from
a random number drawn from ![$[0,1]$](img22.png) then the distance criterion is accepted.
There is also a criterion for the difference in committor probabilities (see PSCALE).
If
then the distance criterion is accepted.
There is also a criterion for the difference in committor probabilities (see PSCALE).
If 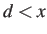 then the distance criterion is always satisfied; above
then the distance criterion is always satisfied; above 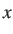 the probability
of acceptance decreases exponentially.
the probability
of acceptance decreases exponentially.
- DUMMYRUN: specifies that no OPTIM jobs should be submitted; the program just
sleeps instead.
- DUMMYTS: specifies that dummy transition state entries should be created for
selected pairs of minima. The corresponding database of minima and dummy transition states
can be refined using any of the usual schemes, e.g. SHORTCUT, FREEPAIRS, etc.
Either BHINTERP or BISECT needs to be specified in the pathdata file,
so that OPTIM jobs only locate new local minima and feed them to PATHSAMPLE
via min.data.info files.
The dummy entries in ts.data include connections for all nearest pairs of minima
for BHINTERP, but only for successive pairs of minima with BISECT.
The dummy energy and log product of mass-weighted Hessian eigenvalues for each
ts.data entry are chosen according to properties of the two minima.
The current energy estimate is the energy of the higher minimum plus the minimum distance
between the two minima.
The estimate for the log product of eigenvalues simply rescales the value for the higher minimum,
effectively removing one degree of freedom.
At some point a decision must be made to start linking the minima via genuine transition
states. To do this we use USEPAIRS, which reads a sequence of stationary points in
the format of the Epath file.
The dummy ts.data and pairs.data files must be renamed or removed.
- DUMPGROUPS: specifies that the groups of potential energy minima and transition
states corresponding to groups of free energy minima and transition states should be dumped
to files minima_groups and ts_groups following regrouping.
- EDIFFTOL x: two stationary points will only be identified as the
same structure if their energy difference is less than x and their minimum
distance is less than the threshold specified by GEOMDIFFTOL.
ETOL specifies the same parameter and is provided for backward compatibility.
- ENERGY x: specifies that the microcanonical ensemble at energy x is to be used
to calculate rate constants. Mutually exclusive with TEMPERATURE.
- ETOL x: energy difference criterion for distinguishing stationary points.
Used with ITOL in PATHSAMPLE.2.0 and GEOMDIFFTOL in
PATHSAMPLE.2.1. EDIFFTOL specifies the same parameter.
- EVCUT x: tolerance for Hessian eigenvalues or frequencies read from
a path.info file by the getallpaths subroutine.
If an eigenvalue is detected that should belong to the positive set by lies below
x (default value
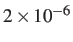 ) the min-sad-min triple is skipped.
) the min-sad-min triple is skipped.
- EXEC a: name of the OPTIM executable to be called by PATHSAMPLE.
- EXTRACTMIN n: extract the coordinates of minimum n from
points.min and write them to file extractmin. If the CHARMM
keyword is present then a pdb format extractedmin.pdb will also be
produced. If
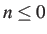 then all the minima are extracted to extractmin.
then all the minima are extracted to extractmin.
- EXTRACTTS n: extract the coordinates of minimum n from
points.ts and write them to file extractts.
If the CHARMM
keyword is present then a pdb format extractedts.pdb will also be
produced. If then all the transition states are extracted to extractts.
- FROMLOWEST (NOLABELS): reads GMIN lowest files and writes PATHSAMPLE points.min
and min.data files. Frequencies are not currently calculated, and so FROMLOWEST
can only be used when NOFRQS is specified. If you GMIN lowest file does NOT contain atom labels
at the start of each line, the NOLABELS option should be used.
- FREEPAIRS x1 x2 x3: connection pairs are chosen based upon the same free energy
regrouping scheme as REGROUPFREE.
x1 corresponds to the free energy barrier height below which free energy
minima are combined.
Free energy minima that lie more than x2 units of energy above the global
free energy minimum group are ignored.
x3 is the energy increment used in the superbasin analysis to estimate
barriers.
Pairs of potential energy minima are chosen from groups based on the ratio
of free energy barrier height to product divided by free energy difference
from product. Hence we highlight groups that correspond to frustration,
with similar energies to the product but high barriers.
Local minima from these free energy groups are then chosen
based upon an additional shortest distance criterion.
Searches are not repeated for the same potential energy minima.
NOTE: for this to work efficiently it is important to have the lowest
minima specified in min.A and DIRECTION set to AB, so that
the A minima are product, and lie at the bottom of the landscape.
See also the PAIRLIST keyword.
- FREEZE n1 n1...: specifies that atoms
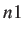 ,
, 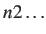 , should be frozen.
This keyword must come after the NATOMS keyword.
, should be frozen.
This keyword must come after the NATOMS keyword.
- FRICTION
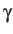 : specifies that rate constants should be
calculated using the multi-dimensional version of Kramer's theory described by
Berezhkovskii, Pollak and Zitserman.[12]
is a phenomenological `friction' coefficient in frequency units, which
must correspond to the units for the Hessian eigenvalue product in the
ts.data file. This formulation requires the imaginary normal mode
frequency for each transition state, and the ts.data file must therefore
contain a ninth column corresponding to the unique negative Hessian eigenvalue.
For the CHARMM and AMBER force fields this eigenvalue is the
angular frequency squared in SI units, while for all other potentials it will
be in reduced units. Such databases can be extended without using the `friction'
formulation by setting the IMFRQ keyword (or by using
: specifies that rate constants should be
calculated using the multi-dimensional version of Kramer's theory described by
Berezhkovskii, Pollak and Zitserman.[12]
is a phenomenological `friction' coefficient in frequency units, which
must correspond to the units for the Hessian eigenvalue product in the
ts.data file. This formulation requires the imaginary normal mode
frequency for each transition state, and the ts.data file must therefore
contain a ninth column corresponding to the unique negative Hessian eigenvalue.
For the CHARMM and AMBER force fields this eigenvalue is the
angular frequency squared in SI units, while for all other potentials it will
be in reduced units. Such databases can be extended without using the `friction'
formulation by setting the IMFRQ keyword (or by using 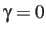 ).
).
- GEOMDIFFTOL x: two stationary points will only be identified as the
same structure if their minimum separation is less than x and their energy
difference is less than the threshold specified by EDIFFTOL.
- GT n1 n2: specifies that the graph transformation approach[4]
should be used to calculate rate constants.
Minima with n1 connections or fewer are iteratively removed before the rate
calculation. n2 specifies that the rates should be calculated every n2 cycles
during the refinement of the stationary point database. For large databases making n2 equal to one
could slow the sampling down significantly.
To run a GT calculation without changing the stationary point database set CYCLES 0.
See keyword GT2 for a more general implementation of graph transformation.
The rates produced from the GT keyword correspond approximately to the
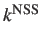 rate constants [4,5].
Branching probabilities for return to the same initial state are set to zero,
and all the other connections are renormalised accordingly. Hence the final branching
probabilities for a given source include arbitrary revisits to that source before
a path reaches a different source or a product, so they do not quite correspond to
committor probabilities. In practice, the resulting rate constants generally seem to
be close to the values.
rate constants [4,5].
Branching probabilities for return to the same initial state are set to zero,
and all the other connections are renormalised accordingly. Hence the final branching
probabilities for a given source include arbitrary revisits to that source before
a path reaches a different source or a product, so they do not quite correspond to
committor probabilities. In practice, the resulting rate constants generally seem to
be close to the values.
- GT2 This keyword has been removed. Please use SGT,
DGT or SDGT.
- GT2RSwitch x : specifies that the generalised graph transformation
routines switch from single to double precision when the density reaches x.
The default value is
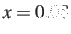 .
See keyword SDGT and subroutine GT2 for more information.
.
See keyword SDGT and subroutine GT2 for more information.
- GT2PTOL x: specifies the threshold for deciding that a node is dead
in the generalised graph transformation routines. The default value is
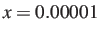 .
.
- IMFRQ: specifies that the ts.data file should contain a ninth
column containing the unique negative Hessian eigenvalue. This
is the eigenvalue corresponding to the imaginary normal mode frequency.
See also the FRICTION keyword, which implies the same format for
ts.data and calculates the rate constants using a phenomenological
description of dynamical solvent friction.
- INTCONSTRAINT intconstrainttol intconstraintdel intconstraintrep intconstrainrepcut
intconsep intrepsep
specifies quasi-continuous interpolation via an auxiliary constraint potential, as
for OPTIM. Note that some of the OPTIM parameters are not needed
for PATHSAMPLE, and are omitted.
In PATHSAMPLE this keyword is only used to define the QCI metric, which can
be used in selecting minima for connection. It is never used with any intervening
images. PATHSAMPLE does not use congeom or congeom.dat files:
it simply calculated the QCI metric based on pairs of local minima.
intconstrainttol is used to determine constrained distances. The deviation
of the distance between a given pair of atoms
in all reference structures from the average must be less than
intconstrainttol, default 0.1, for this separation to constitute a constraint.
If a percolating network of constraints does not result then intconstrainttol
is increased by 10% and the analysis repeated.
intconstraintdel multiplies the constraint penalty term in the auxiliary potential,
default 10.0.
intconstraintrep multiplies the repulsive penalty term in the auxiliary potential,
default 100.0.
intconstrainrepcut is the cutoff for the repulsive penalty term in the auxiliary potential,
default 1.7.
intconsep is the maximum difference between the order number of atoms for
which constraints are allowed. The default is 15, which seems appropriate for
biomolecules. However, if a congeom file of reference minima is specified, or
a corresponding congeom.dat file is present, then intconsep can be set
greater than the total number of atoms.
intrepsep is the minimum difference in order number of atoms for which repulsive
terms are added to the potential. The default is zero; there are no repulsive terms
between constrained atoms.
- ITOL x: principal moment of inertia tensor difference criterion for
distinguishing stationary points. All three values are compared for stationary points
in the initial setup phase. However, unlike PATHSAMPLE.2.0, the alternative
criterion specified by GEOMDIFFTOL is used to distinguish stationary points
in PATHSAMPLE.2.1.
- JOBSPERNODE jpn: specifies the number of jobs to run on each core for a
calculation using a scheduler such as PBS to send jobs to distributed nodes. The cores
must be defined by the job submission script in the nodes.info file.
The keyword is not called JOBSPERCORE historical reasons and backward compatibility.
Unless you really want to run more than one job per available core at one time then
jpn can be omitted: the default value is one. The PBS keyword should
probably be used instead of JOBSPERNODE going forward.
- KMC n1 x n2: perform n1 KMC runs having iteratively removed
minima with n2 connections or fewer.
x specifies a pair threshold for the product, p, of forward and backward
branching probabilities between two minima.
If p is greater than x then a leapfrog move will be attempted to a
different connected minimum when either of the pair is encountered.
Setting p to one or more means that leapfrog moves will not be used.
No additional stationary points will be calculated in a KMC
run: the CYCLES keyword is ignored if it is present in odata.
Only one of the GT, GT2, KMC and KMCcommit keywords should be
present in a given pathdata file.
- KMCcommit n1 x1 x2 x3 x4 n2: specifies a rate constant calculation using
committor probabilities and appropriate waiting times. This subroutine can calculate
both `SS' and `NSS'-type rate constants.[4]
n1 is the number of KMC trajectories to run. If n1 is zero then
the waiting time in a given minimum is the the escape time
to its connected neighbours. Combined with the committor probabilities,
these waiting times give `SS' rate constants. If n1 is greater than zero then
the waiting times are calculated for KMC runs that terminate as soon as they reach
a minimum in either the A or the B region. The resulting rate constants
are described as `NSS'.[4]
x1 is the maximum number of iterations allowed in the committor probability
calculation; it is a real number because the value required may exceed the size allowed
for an integer.
x2 is a convergence condition for the committor probabilities, and is defined
in terms of the percentage change between iterations.
x3 is the
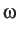 parameter used in the successive overrelaxation calculation
of the committor probabilities. It should be greater than or equal to one, but less than 2.
The default value is one, which results in Gauss-Seidel iterations.
x4 specifies a pair threshold for the product, p, of forward and backward
branching probabilities between two minima.
If p is greater than x then a leapfrog move will be attempted to a
different connected minimum when either of the pair is encountered.
Setting p to one or more means that leapfrog moves will not be used.
n2 determines the minimum connectivity allowed for local minima to be
admitted into the calculation. The minima are iteratively pruned to remove those
with n2 connections or fewer.
No additional stationary points will be calculated in a KMCcommit
run: the CYCLES keyword is ignored if it is present in odata.
Only one of the GT, GT2, KMC and KMCcommit keywords should be
present in a given pathdata file.
parameter used in the successive overrelaxation calculation
of the committor probabilities. It should be greater than or equal to one, but less than 2.
The default value is one, which results in Gauss-Seidel iterations.
x4 specifies a pair threshold for the product, p, of forward and backward
branching probabilities between two minima.
If p is greater than x then a leapfrog move will be attempted to a
different connected minimum when either of the pair is encountered.
Setting p to one or more means that leapfrog moves will not be used.
n2 determines the minimum connectivity allowed for local minima to be
admitted into the calculation. The minima are iteratively pruned to remove those
with n2 connections or fewer.
No additional stationary points will be calculated in a KMCcommit
run: the CYCLES keyword is ignored if it is present in odata.
Only one of the GT, GT2, KMC and KMCcommit keywords should be
present in a given pathdata file.
- KSHORTESTPATHS npaths nconnmin extraprinting: calculate the npaths paths
with the largest contributions to the rate constant when intervening minima are
put in steady state. Coded by Dr Joanne Carr.
nconnmin specifies that minima with nconnmin connections or fewer
should be disregarded in this analysis. Setting the optional argument extraprinting to "T"
turns on extra output printing for each path (off by default).
- LPERMDIST n thresh cut: is the same local
permutational alignment procedure as for OPTIM.
n is the maximum number of extra atoms to include in running
minperm for each permutable group (default 4), rather than
treating the whole system at the same time, as for PERMDIST.
The extra atoms that define the neighbourhood of the permutable group are
added one at a time in order of their distance from the centre of coordinates of the
group, averaged over the start and finish geometries.
If the optimal aligned distance is greater than thresh (default value 0.2)
then the atom is not included in the neighbour list.
This procedure continues until there are n atoms in the neighbour list,
or no more atoms within the cutoff distance cut, default 10.0.
This keyword requires the auxiliary file perm.allow to specify permutable atoms, otherwise
all atoms are assumed to be permutable. The absence of a perm.allow
file is considered a mistake for CHARMM runs.
- LOWESTFRQ: specifies that the lowest positive eigenvalues associated
with the Hessian and mass-weighted Hessian should be read from min.data.info
files and recorded in min.data.
These eigenvalues can then be used in combination with DUMMYTS
to estimate the energy and frequency factor associated with dummy transition states.
The LOWESTFRQ keyword must also appear in odata.bhinterp or odata.bisect.
The current estimate for dummy transition state parameters does not require these
extra data.
- MACHINE: if specified, certain files are written and read as
binary rather than plain text.
- MACROCYCLE n: sets up permutational isomers for a
macrocyclic system. The number of repeat units in the macrocycle is given by
n (e.g. n=4 for cyclo-[Gly
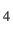 ]). The order of the atoms in
each repeat unit must be the same and these permutations should not be added to
the perm.allow file. This keyword has only been tested on cyclic peptides using
the AMBER potential.
]). The order of the atoms in
each repeat unit must be the same and these permutations should not be added to
the perm.allow file. This keyword has only been tested on cyclic peptides using
the AMBER potential.
- MAKEPAIRS pairfile: use in conjunction with ADDMIN or READMIN to produce an Epath-style pairfile which can then be used with USEPAIRS to connect a chain of minima together. The minima from the min.data.info are read into the PATHSAMPLE database as normal, but in addition the pairfile records the indices of the minima in the order in which they appeared in min.data.info. Running PATHSAMPLE with USEPAIRS then connects every pair of minima from the original min.data.info file, in the order they appeared. Minima may appear multiple times in the same pairfile if they appear multiple times in min.data.info. The exception is consecutive duplicate minima, which are omitted.
- MAXBARRIER x: use in the post-processing analyses to set a criterion for excluding transition
states if the barriers from both connected minima exceed x. The threshold x is reset to infinity
on converting potential energies to free energies during a run.
- MAXDOWNBARRIER x: specifies a maximum downhill barrier
of x for DIJKSTRA analysis.
The downhill barriers for the path with the largest contribution to the
non-recrossing steady-state rate constant are sorted and those that exceed
x are analysed further. The transition state in question is removed
and the DIJKSTRA analysis performed again if a connected path is still possible.
If a connected path is no longer possible the transition state in question is
reinstated. A list of `shiftable' transition states is provided at the end.
Note that the order in which transition states are removed could affect this list,
so that other combinations of `shiftable' transition states may be possible.
The downhill direction can only be defined locally once the complete
DIJKSTRA path is known, since `downhill' is determined by the energies
of the two endpoints.
- MAXTSENERGY x: specifies that transition states above energy should
not be included in the database. Probably most useful with CHARMM, where genuine
transition states with silly energies can certainly occur and give rise to underflow.
This keyword performs the same function as TSTHRESH, and is provided because
OPTIM uses MAXTSENERGY.
- MERGEDB dir: merge the min.data, min.ts, points.min,
points.ts, min.A and min.B files from directory dir with
those in the current directory. Common stationary points are identified and
not repeated. Merged results are created corresponding to all six of the above files.
- MINBARRIER x: use in the post-processing analyses to set a criterion for excluding a transition
state if the barrier in either direction is lower than x. Designed to allow exclusion of TSs
present in the database that lie lower in potential energy than the connected minima, by setting x to zero.
The default value is (basically) minus infinity. Note that a transition state may legitimately lie lower
than the connected minima in free energy.
- NATOMS n: the number of atoms in the system; must be present.
- NCONNMIN n: specifies that minima with n connections or fewer
should be disregarded in regrouping and rate calculations. This parameter
can be set via other keywords, but NCONNMIN is provided for use with
SGT, DGT, SDGT and also PFOLD. The connection condition is evaluated recursively.
- NEWCONNECTIONS n maxattempts firstmin:
this keyword provides a fully parallelised way to grow a database using single-ended
transition state searches.
It therefore provides an alternative to procedures such as SHORTCUT and
UNTRAP.
Starting from firstmin (default 1) PATHSAMPLE will step through all the
known minima and perform up to maxattempts transition state and pathway
searches (default 10) according to the template odata.tspath file, which must be
present in the working directory.
The next minimum will be considered if the current minimum has
n or more connections (default 0).
for which the default is 10.
The odata.tspath file needs to contain OPTIM PATH
and DUMPALLPATHS directives
so that a path.info file is created.
- NGT nconnmin disconnectsources density size compressedrowswitch: specifies that the new
graph transformation approach should be used to calculate rate constants.
Minima with nconnmin connections or fewer are iteratively removed before the rate calculation.
This calculation produces
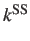 , and for both
, and for both
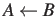 and
and 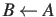 , as well as committor probabilities.
If disconnectsources is true (default is false) then is also
calculated. For single sources is the same as .
The algorithm switches from sparse to dense storage when the number of connections
exceeds density times the maximum possible value, with the restriction that
there must be size minima or fewer remaining. The default values for density
and size are 0.3 and 11000. Within the initial sparse storage scheme, the algorithm will use
compressed row (vector) storage instead of full, rectangular matrix storage if the memory required for the
two rectangular matrices at the start of the NGT calculation would exceed compressedrowswitch Gb.
See also keyword GT2 for the original graph transformation calculation of .
The committor probabilities for the end point minima, which are obtained on removing all the
intervening minima, are written to two files: commit.ngt.AB for direction B to A and commit.ngt.BA for the opposite
direction. The entries for the intervening minima are zeroes. These files have the same form and ordering as commit.data.
The PFOLD keyword (see below) may be given in addition to NGT, in which case a commmittor probability
calculation using the specified control parameters will immediately follow the NGT calculation, and
will be seeded with the known (and fixed) committor probabilities from NGT for the end point minima.
A file commit.data, with one entry for each minimum in the pre-resorting order, will be written at the
end of this calculation containing the committor probabilities for the direction specified via DIRECTION.
, as well as committor probabilities.
If disconnectsources is true (default is false) then is also
calculated. For single sources is the same as .
The algorithm switches from sparse to dense storage when the number of connections
exceeds density times the maximum possible value, with the restriction that
there must be size minima or fewer remaining. The default values for density
and size are 0.3 and 11000. Within the initial sparse storage scheme, the algorithm will use
compressed row (vector) storage instead of full, rectangular matrix storage if the memory required for the
two rectangular matrices at the start of the NGT calculation would exceed compressedrowswitch Gb.
See also keyword GT2 for the original graph transformation calculation of .
The committor probabilities for the end point minima, which are obtained on removing all the
intervening minima, are written to two files: commit.ngt.AB for direction B to A and commit.ngt.BA for the opposite
direction. The entries for the intervening minima are zeroes. These files have the same form and ordering as commit.data.
The PFOLD keyword (see below) may be given in addition to NGT, in which case a commmittor probability
calculation using the specified control parameters will immediately follow the NGT calculation, and
will be seeded with the known (and fixed) committor probabilities from NGT for the end point minima.
A file commit.data, with one entry for each minimum in the pre-resorting order, will be written at the
end of this calculation containing the committor probabilities for the direction specified via DIRECTION.
- NINTS n: the number of internal coordinates; required for runs
with the UNRES potential.
- NIMET NIHEAM7 : these keywords ensure that the EAM4 and EAM7 potentials for hydrogen on a nickel surface
(see OPTIM manual for more details) get the right atom symbols when called from pathsample.
- NOFRQS: specifies that frequencies are not present in path.info files.
Used for very large systems where frequency calculations are not feasible in conjunction
with the same OPTIM keyword.
The log product of positive eigenvalues is set to unity where it is read in from
ts.data and min.data in setup and Dijsktra for consistency.
- NOINVERSION: turns off inversion of structures for distance
metric in minpermdist.
- NTIP 4: calls the TIP4P model of water molecules.
- OHCELL : allow point group operations for a cubic
supercell in subroutine minpermdist.f90 permutational distance minimisation.
- ORBITGEOMTOL x: sets the cutoff value for assigning atoms to the
same orbit when analysing the standard orientation in routine myorient
when LPERMDIST is specified.
The default value for x is 0.3.
If x is too small it is possible for permutational isomers to be missed.
- ORDER x: reads in an order parameter threshold. Currently not used.
- PAHA n: calls a finite system of polycyclic aromatic hydrocarbons (PAH) interacting
via a general anisotropic potential developed from first principles [13]. The PAH ID
n defines the PAH molecule: 1 for benzene is the only option currently available.
- PAIRLIST n: specifies that the list of minima for subsequent connection attempts
should be regenerated every n cycles. The default is for the list to remain
fixed throughout the run unless we run out of candidates. This option has been
implemented for the FREEPAIRS, SHORTCUT, UNTRAP and
DIJPAIR keywords and for the default connection
strategy based on differences in the committor probability. It has not been
implemented for the CONNECTREGION keyword.
- PBS jpn: specifies the number of jobs to run on each core for a
calculation using a scheduler such as PBS to run OPTIM jobs on
distributed nodes. The cores
must be defined by the job submission script in the nodes.info file.
Unless you really want to run more than one job per available core at a time then
jpn can be omitted: the default value is one.
- PERMDIST x: specifies that all distance metrics should include minimisation
with respect to permutational isomerisation, as well as optimal orientation.
Should be used with the same keyword in OPTIM.
Requires the auxiliary file perm.allow to specify permutable atoms.
The first line of the perm.allow file must contain an integer
that specifies the number of primary groups of interchangeable atoms.
The groups then follow, each one introduced by a line with two integers
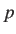 and
and 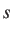 that specify the number of permutable atoms in the primary group and the number of other sets
of permutable atoms associated with the primary set.
may be zero.
Each secondary set of permutable atoms has members.
The following line contains the indices of the permutable atoms
in the primary set and then
the indices of the atoms in each of the secondary sets, one set at
a time.
The parameter x is the distance tolerance for assigning atoms to orbits
in the myorient standard orientation routine.
If x is too small it is possible for permutational isomers to be missed.
that specify the number of permutable atoms in the primary group and the number of other sets
of permutable atoms associated with the primary set.
may be zero.
Each secondary set of permutable atoms has members.
The following line contains the indices of the permutable atoms
in the primary set and then
the indices of the atoms in each of the secondary sets, one set at
a time.
The parameter x is the distance tolerance for assigning atoms to orbits
in the myorient standard orientation routine.
If x is too small it is possible for permutational isomers to be missed.
For the phenylalanine example illustrated above we must allow three other pairs of
atoms to exchange if we swap 7 and 8. Hence a suitable perm.allow entry is
1
2 3
7 8 5 6 1 2 3 4
Here 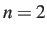 and
and 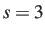 : if we exchange 7 and 8 then we must also exchange 5 and 6,
1 and 2, and 3 and 4. There are two atoms in each of the three secondary sets,
since we have specified 7 and 8 as the two primary atoms.
: if we exchange 7 and 8 then we must also exchange 5 and 6,
1 and 2, and 3 and 4. There are two atoms in each of the three secondary sets,
since we have specified 7 and 8 as the two primary atoms.
Here is an example perm.allow file for a water trimer using
the flexible QTIP4PF potential, where the energy is invariant to permutations
of water molecules and to exchanges of hydrogens in the same molecule. However,
hydrogens cannot exchange between different oxygens:
4
3 2
1 4 7 2 3 5 6 8 9
2 0
2 3
2 0
5 6
2 0
8 9
The first group of three oxygens has two atoms that must move with each oxygen,
i.e. atoms 2 and 3 for oxygen 1, etc. Hydrogen permutations for each oxygen are
allowed by the three following groups. This scheme allows atoms to appear in more
than one group. There must be a group containing each complete set of permutations
in order for permutation-inversion isomers to be recognised. The format
is compatible with an older scheme, where only pair swaps were allowed for
associated atoms, but now allows for more general permutations.
Scripts to generate allowed permutations automatically for CHARMM and AMBER are available from
the group web site. It is essential to use symmetrised versions of the corresponding
force fields!
- PERMISOMER: equivalent to PERMDIST above, expect that
distance minimisation
with respect to permutational isomerisation is only carried out for stationary
points with energies that differ by less than the parameter specified by EDIFFTOL.
Hence permutational isomers should be identified, but the distance between minima with
different energies is not minimised with respect to atom permutations.
This option is provided for cases where the number of permutations is too
minimise distances with respect to permutations in general.
Should be used with the PERMDIST keyword in OPTIM.
The auxiliary file perm.allow specifies which atoms may be permuted; see
PERMDIST above.
Note that the NATOMS keyword must precede PERMISOMER in the
pathdata file.
- PERSIST einc pethresh peqthresh perthresh :
Persistence analysis along the lines of Cazals et al.
We perform a persistence analysis using superbasins at energy increments of einc
up to a maximum energy range of pethresh.
This analysis uses the persistence for each minimum, defined as the barrier to the transition state
where that minimum can access a lower-energy minimum.
Output files are min.persist, min.persist.select and min.include. The latter two
include only some of the most persistent minima, selected based on a lower bound
equilibrium occupation probability peqthresh and a lower bound, perthresh, on the barrier
height defined by the persistence (actually the deviation from the diagonal in a
persistence diagram, which is the same aside from a factor of
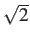 ). The usual criteria for transition
states and the connectivity of minima also apply.
The file min.include has a format suitable for reading via the
PICK and IDMINFILE keywords in disconnectionDPS. The format of the min.persist* files
is: rank (ordered by persistence), energy, energy+barrier, barrier(=persistence), index of the minimum,
deviation from the diagonal. See also PERSISTEXACT
). The usual criteria for transition
states and the connectivity of minima also apply.
The file min.include has a format suitable for reading via the
PICK and IDMINFILE keywords in disconnectionDPS. The format of the min.persist* files
is: rank (ordered by persistence), energy, energy+barrier, barrier(=persistence), index of the minimum,
deviation from the diagonal. See also PERSISTEXACT
- PERSISTEXACT ALL:
For use with PERSIST; specifies that the barrier heights used to define the persistences
should be obtained via an efficient exact method, rather than the approximate method using
a superbasin analysis. Note that the values of einc and pethresh from PERSIST are not
used here, but must still be present if you want to specify values for peqthresh and perthresh.
The optional argument ALL instructs the program to report persistences for
all the valid minima in the network; the default is to report values only for the minima in the same connected
component as the global minimum (which matches the situation for the approximate method).
- PERTURB x: initial maximum perturbation of the Cartesian coordinates of a minimum,
when looking for a new connected minimum via a single-ended transition state search.
- PFOLD n1 n2 x PFOLDCONV: provides control parameters for the calculation
of committor probabilities for local minima. The calculation
may be performed on-the-fly during the construction of a stationary point database (if used together
with CYCLES <non-zero value>) or as a post-processing analysis (CYCLES 0).
All new local minima are assigned an initial value of zero.
If a file commit.data exists in the working directory then initial values at the start of the run
will be taken from there.
n1 iterations are performed every n2 cycles with parameter in
the successive overrelaxation equal to x.
PFOLDCONV is the threshold on the largest fractional difference between Pfold values at
consecutive iterations for early convergence of the algorithm (before the full n1 iterations
have been performed). The default value is 0.0001.
Note that a valid solution also exists for all committor probabilities equal to unity.
I have seen the iteration converge to this solution for a very small database that
has just been created with DIJINIT.
A file commit.data, with one entry for each minimum under consideration in the pre-resorting order,
will be written at the end of a
committor probability calculation for the pathway direction specified in DIRECTION.
The PFOLD keyword may be given in addition to NGT, in which case a commmittor probability
calculation using the specified control parameters will immediately follow the NGT calculation, and
will be seeded with the known (and fixed) committor probabilities from NGT for the end point minima.
- PLANCK h: specifies the value of Planck's constant in units of the
prevailing energy unit times seconds. This numerical value is required for
regrouping free energies. For example, for the CHARMM potential needs
to be
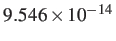 skcal/mol
See §7 for more details.
skcal/mol
See §7 for more details.
- PULL : add this keyword if the system has a non-zero static force
added to the potential. PATHSAMPLE.2.1 only uses this keyword to determine the
number of zero Hessian eigenvalues (four). The attached atoms and force magnitude
are not required.
- PSCALE x: determines how the difference in committor probabilities,
, is used to select the pair of minima used in the next attempted connection. If
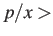 a random number drawn from then the distance criterion is accepted.
There is also a criterion for the minimum distance (see DSCALE).
If
a random number drawn from then the distance criterion is accepted.
There is also a criterion for the minimum distance (see DSCALE).
If 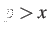 then the committor probability criterion is always satisfied; below the probability
of acceptance decreases linearly.
then the committor probability criterion is always satisfied; below the probability
of acceptance decreases linearly.
- RANDOMMETRIC n: for DIJINITCONT jobs this keyword limits
the number of metric calculations for each new minimum to n. The n
other minima are chosen randomly. The cost of adding each new minimum to the
database is therefore constant, rather than growing linearly with the number
of minima in the database. If n is less than the max
parameter for DIJINITCONT then the metric values are padded with
enormous values.
- RANROT n: for use with PERMDIST. Specifies
that n random orientations will be tried as well as the
initial standard alignment in minpermdist.f90. Several hundred
random orientations may be needed to find the best translational/rotational/permutational
alignment of end points.
- RATESCYCLE t1 t2
 tn : calculate rate constants
at the end of every cycle for temperatures t1, t2, etc.
Each temperature and forward and backward rate constants are dumped to
file NGT.rates.new.
Any number of temperatures can be specified, so long as continuation
lines are used if the line length exceeds 80 characters.
This keyword was introduced to make the LJ
tn : calculate rate constants
at the end of every cycle for temperatures t1, t2, etc.
Each temperature and forward and backward rate constants are dumped to
file NGT.rates.new.
Any number of temperatures can be specified, so long as continuation
lines are used if the line length exceeds 80 characters.
This keyword was introduced to make the LJ demonstration cleaner.
demonstration cleaner.
- RBAA: specifies the number of rigid bodies being considered in the angle-axis. NB: this is not part of the GENRIGID framework.
- RBSYM: specifies that internal symmetry operations permuting equivalent sites for
rigid-body building blocks exist. The operations are read in from a file rbsymops, which must
exist in the working directory. The first line of the file is an integer that specifies the number
of operations generated by proper axes of rotation including the identity operation; the operations
are then read in one at a time in a representation consisting of a unit vector, defining the
axis in the reference frame, and an angle in degrees, describing the magnitude of rotation about
that axis. The first operation has to correspond to the identity operation.
This keyword has to be combined with PERMDIST so that structures are aligned and
a distance metric considered based on permutations of identical rigid
bodies and any internal symmetry operation of each rigid body that is a symmetry
of the potential energy function. NB: this is not part of the GENRIGID framework.
- READMIN file: reads in the data for one or more minima from file
in OPTIM min.data.info format and then stops.
Any number of min.data.info files can be concatenated into file.
New entries will be created in min.data and points.min.
If min.A and min.B do not exist they are not created.
READMIN therefore provides an alternative way to start an initial
path calculation by providing minima, so long as min.A and min.B
are created by hand.
It also provides a way to add additional minima to an existing database, since
new entries should be appended. There is a check to make sure that the minima
added are different from all those currently known.
- REGROUP x: regroup the A and B sets using a disconnectivity graph
analysis [14,15,2] at energy x.
Any intervening minima that share a superbasin with an A or B minimum are reclassified
to belong to the A and B sets, respectively.[4]
The number of minima and transition states does not change: only the
classification of minima as A or B is affected,
which will change any rate analysis that follows.
The corresponding
minima are printed in files min.A.regrouped and min.B.regrouped.
- REGROUPFREE x: the database will be regrouped by combining free
energy minima where forward and backward free energy barriers are both
less than x. Regrouping is performed iteratively until no further free
energy minima merge.
In this scheme, groups that contain product and reactant minima
(A and B sets) are not allowed to merge.
The regrouping continues even if product and reactant groups
could merge according to the barrier threshold: such mergers are simply forbidden.
Don't forget to assign an appropriate value for the Planck constant using the
PLANCK keyword.
If one of the DIJKSTRA, SDGT, SGT, or DGT keywords is
present the corresponding analysis will be performed using the new groups
and free energies.
- REGROUPFREEAB x: the database will be regrouped as for
REGROUPFREE above, but only the A and B groups are changed.
- REGROUPPE x: the database will be regrouped based upon
a superbasin analysis at potential energy .
Minima will be reassigned as A and B-type if they are in a superbasin
with an A or B minimum for this threshold.
New files min.data.regrouped, ts.data.regrouped, min.A.regrouped
and min.B.regrouped will be created.
Don't forget to assign an appropriate value for the Planck constant using the
PLANCK keyword. See also REGROUPRATE.
REGROUPPE and REGROUPRATE are different from REGROUP
in that they actually change the internal database structure into free energy
minima and groups of transition states linking them.
In this scheme, groups that contain product and reactant minima
(A and B sets) are not allowed to merge.
The regrouping continues even if product and reactant groups
could merge according to the barrier threshold: such mergers are simply forbidden.
If one of the DIJKSTRA, SDGT, SGT, or DGT keywords is
present the corresponding analysis will be performed using the new groups
and free energies.
- REGROUPPERSIST thresh :
regrouping analogous to REGROUPFREE except that minima in groups defined
by the min.include file (which contains some of the most persistent minima, see
PERSIST) are also not allowed to merge, like the A and B
sets (unless ALLOWAB is set). The PERSIST keyword must also be present
(though the code could easily be changed to use any pre-existing min.include file...).
- REGROUPRATE x: the database will be regrouped based upon harmonic transition
state theory rate constants. Minima are in the same group if they can interconvert
via a path that involves only rate constants greater than .
Minima will be reassigned as A and B-type if they are in a group
with an A or B minimum for this threshold.
New files min.data.regrouped, ts.data.regrouped, min.A.regrouped
and min.B.regrouped will be created.
Don't forget to assign an appropriate value for the Planck constant using the
PLANCK keyword. See also REGROUPPE.
REGROUPPE and REGROUPRATE are different from REGROUP
in that they actually change the internal database structure into free energy
minima and groups of transition states linking them.
In this scheme, groups that contain product and reactant minima
(A and B sets) are not allowed to merge.
The regrouping continues even if product and reactant groups
could merge according to the barrier threshold: such mergers are simply forbidden.
If one of the DIJKSTRA, SDGT, SGT, or DGT keywords is
present the corresponding analysis will be performed using the new groups
and free energies.
- REMOVESP: the minima specified in files min.remove and
ts.remove are removed and new files min.data.removed,
ts.data.removed, min.A.removed, min.B.removed,
points.min.removed and points.ts.removed
are created with a consistent numbering scheme for the remaining stationary
points. The first lines of min.remove and
ts.remove must contain a single integer, which is the number of
stationary points listed for removal on the remaining lines.
- REMOVEUNCONNECTED set : the database is rewritten to files
min.data.removed, points.min.removed, etc.
Minima disconnected from set, where set is A B or AB,
are removed, along with all transition states that connect them.
Minima with NCONNMIN connections or fewer are also removed (this
condition is applied recursively). The default for set is AB, which will remove
minima that have no connection to any member of the A or B sets.
- RETAINSP: minima not specified in file min.retain
are removed and new files min.data.retained,
ts.data.retained, min.A.retained, min.B.retained,
points.min.retained and points.ts.retained
are created with a consistent numbering scheme for the remaining stationary
points. The first line of min.retain
must contain a single integer, which is the number of
minima listed on the remaining lines.
Transition states are only retained if they link two minima specified in the
min.retain file.
- REWEIGHT nrwbins nrwreactant energyfile:
conditional probabilities of reactant minima are obtained from the quench
probabilities calculated using the energies in file energyfile.
These energies could come from systematic quenching from an MD trajectory at
the same temperature, for example.
nrwbins is the number of bins to use in representing the quench
probability distribution and nrwreactant is the number of bins
to use in representing the minima in the reactant set.
This is a somewhat experimental option. Consult DJW before using!
- RELATIVEE: subtract the potential energy of the global minimum
from all the minima and transition states, as well as the treshold for
transition state neglect. This shift of the energy origin may
help to avoid some numerical underflow or overflow issues in rate
and therodynamics calculations, especially at
low temperature.
It can only be used in post-processing, since we shift only the potential energy
values that are read in initially.
- RFMULTI tau Tlow Thigh Tinc steps pfshift:
run the REGROUPFREE analysis as a function of temperature for a fixed
regrouping threshold. The threshold is determined from the observation time scale, tau
and the temperature range is Tlow to Thigh in steps
steps.
A fixed shift for the natural log of the partition functions can be specified
by the parameter pfshift, which may be needed to prevent underflow or overflow.
- RIGIDBODIES n: specifies the number of rigid bodies; only one
of NATOMS and RIGIDBODIES should be present. NB: this is not part of the GENRIGID framework.
- RIGIDINIT n: specifies the use of the generalised rigid body framework. You must also specify
the number of degrees of freedom in the rigidified system, n, so that PATHSAMPLE knows how many
eigenvalues to expect in OPTIM output.
- SDGT DisconnectSources AltPbb Rescale Normalise:
graph transformation rate calculation, which switches from sparse optimised to
dense optimised algorithms when the average number of connections
exceeds a certain threshold [4].
The four arguments are all logicals, so an example input line might look
SDGT T T F F. If true (the default) DisconnectSources specifies that
a full transformation should be performed for each source, disconnecting the
other sources. The resulting rate constants correspond to the KMC result and
[5].
If AltPbb is true (default is false) then additional work is done to
maintain precision, which roughly doubles the execution time, but may be needed at low
temperature.
If Rescale is true (default false) an alternative strategy is used
to try and prevent error propagation.
Setting Normalise true (default false) instructs GT2input.f90 to check the normalisation
of branch probabilities. This should not be necessary.
- SEED n: random number seed.
- SGT DisconnectSources AltPbb Rescale Normalise:
sparse optimised graph transformation rate calculation [4].
The four arguments are all logicals, so an example input line might look
SGT T T F F. If true (the default) DisconnectSources specifies that
a full transformation should be performed for each source, disconnecting the
other sources. The resulting rate constants correspond to the KMC result and
[5].
If AltPbb is true (default is false) then additional work is done to
maintain precision, which roughly doubles the execution time, but may be needed at low
temperature.
If Rescale is true (default false) an alternative strategy is used
to try and prevent error propagation.
Setting Normalise true (default false) instructs GT2input.f90 to check the normalisation
of branch probabilities. This should not be necessary.
- SHORTCUT minsep string: try connecting minima that are further apart than minsep
steps on the best path, as calculated using Dijkstra's algorithm.
In the Dijkstra analysis minima with zero connections are removed.
This value can be changed using a DIJKSTRA n line in pathdata.
If the option argument string is set to BARRIER or RATE then
the strategy is different. We now look for pairs of minima on either side of
the highest barriers or minima linked by the smallest
rate constant on the best path, sorted according to the barrier height or the rate.
minsep is then used to define the largest number of steps away from
the transition states for connection attempts.
- SKIPPAIRS : do not attempt to connect a pair of minima if they are already connected. This is used only with USEPAIRS. Pairs which are rejected on these grounds DO count towards the total number of connections provided by the CYCLES keyword, however the debug printing doesn't recognise that at the present time.
- SLEEPTIME x : set the time used in the system sleep calls
between submission of OPTIM jobs. The default value is 1s. A nonzero
value is needed for small systems on distributed memory machines to prevent
us from running out of ports. A value of zero on a shared memory system can
speed up execution significantly for small systems.
- SSH : OPTIM jobs are submitted to nodes using ssh rather than rsh (default)
- ST : calls a finite system of Stockmayer particles.
- STARTFROMPATH afile n1 n2 : reads an initial path from file afile. The files min.A and min.B are created: min.A contains the entries
1 and n1 and min.B contains the entries 1 and n2.
If min.A already exists then the run will terminate with a warning message
without overwriting it.
- SYSTEM a: the atom type label to be written to odata files for OPTIM,
thus specifying the potential that OPTIM will use.
- TAG n x: atom number n in the system is `tagged' by giving it
an artificial mass x.
- TEMPERATURE x: specifies that the canonical ensemble at reduced temperature x
is to be used to calculate rate constants. Mutually exclusive with ENERGY.
- TRAP: specifies a trapped ion potential. This keyword is needed to set the
number of zero eigenvalues. See the coresponding keyword in OPTIM for more information.
- TSTHRESH x: specifies that transition states above energy should
not be included in the database. Probably most useful with CHARMM, where genuine
transition states with silly energies can certainly occur and give rise to underflow.
- TWOD: the system is two-dimensional.
- UNRES: specifies the UNRES potential.[16]
- UNTRAP einc thresh edeltamin elowbar ehighbar: try connecting minima to eliminate traps.
Connections are attempted between all minima and any of the minima in the
product set based on the largest barriers in the product direction, as estimated
from disconnectivity analysis at energy intervals of
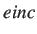 , starting from the
global minimum.
The product basin is defined as the set at the highest energy for which the
minimum in question is not a member.
Candidate pairs of minima are sorted based upon the ratio of the
potential energy barrier to the potential energy difference between the minimum
in question and the lowest product minimum.
Hence this scheme is analogous to the FREEPAIRS procedure, but
uses individual minima and potential energy, instead of free energy.
The original product minimum may be replaced by a minimum from
the same superbasin that is closer to the target minimum, so long as it
lies within an energy
, starting from the
global minimum.
The product basin is defined as the set at the highest energy for which the
minimum in question is not a member.
Candidate pairs of minima are sorted based upon the ratio of the
potential energy barrier to the potential energy difference between the minimum
in question and the lowest product minimum.
Hence this scheme is analogous to the FREEPAIRS procedure, but
uses individual minima and potential energy, instead of free energy.
The original product minimum may be replaced by a minimum from
the same superbasin that is closer to the target minimum, so long as it
lies within an energy 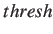 above the minimum to be untrapped.
Minima with zero connections are not considered;
this value can be changed using a DIJKSTRA n line in pathdata, even
though a Dijkstra analysis is not used.
See also the PAIRLIST keyword.
The UNTRAP keyword may provide the best way to remove artificial
frustration from potential energy disconnectivity graphs.
It may be helpful to choose a larger value of einc than would be
used for the superbasin analysis in visualising the disconnectivity graph.
This will encourage connection attempts with minima of lower energy in the
expanded product superbasin.
The optional arguments edeltamin and elowbar are useful for databases
without a well-defined global minimum. If the potential energy difference between the minimum
in question and the lowest product minimum is less than edeltamin, the value of
edeltamin is substituted for this difference. This substitution prevents
connection attempts to minima connected by low barriers but with a very small
energy difference to the lowest product minimum. If the barrier from the lowest product minimum to the minimum in question is less than elowbar, a value of TINY(1.0D0) is given to the barrier instead. This substitution prevents connection attempts to minima connected
by low barriers.
The fifth optional argument ehighbar aids with the selection of minima for
untrapping if you wish to target particular regions. Minima with a barrier height (in the direction
of the lowest-energy product minimum) larger than EHIGHBAR will not be selected.
above the minimum to be untrapped.
Minima with zero connections are not considered;
this value can be changed using a DIJKSTRA n line in pathdata, even
though a Dijkstra analysis is not used.
See also the PAIRLIST keyword.
The UNTRAP keyword may provide the best way to remove artificial
frustration from potential energy disconnectivity graphs.
It may be helpful to choose a larger value of einc than would be
used for the superbasin analysis in visualising the disconnectivity graph.
This will encourage connection attempts with minima of lower energy in the
expanded product superbasin.
The optional arguments edeltamin and elowbar are useful for databases
without a well-defined global minimum. If the potential energy difference between the minimum
in question and the lowest product minimum is less than edeltamin, the value of
edeltamin is substituted for this difference. This substitution prevents
connection attempts to minima connected by low barriers but with a very small
energy difference to the lowest product minimum. If the barrier from the lowest product minimum to the minimum in question is less than elowbar, a value of TINY(1.0D0) is given to the barrier instead. This substitution prevents connection attempts to minima connected
by low barriers.
The fifth optional argument ehighbar aids with the selection of minima for
untrapping if you wish to target particular regions. Minima with a barrier height (in the direction
of the lowest-energy product minimum) larger than EHIGHBAR will not be selected.
Sometimes there are large groups of minima separated by small barriers that form a trap. We only need to find a lower-energy path for one of these minima and untrapping all of them may be a wasted effort. Running disconnectionDPS with the keyword BASINT followed by a particular energy gives the results of a basin analysis at this energy in a file `basins'. If this file is renamed as `noduplicatebasins' and placed in the directory where the PATHSAMPLE job is running, untrap will only pick one minimum from this group for untrapping.
- UNTRAPMETRIC nitems nmatrix baildist: to be used with UNTRAP. nitems is the number of metrics to use (1,2 or 3), nmatrix is the number of closest minima for which distances are calculated (default 2000) and baildist is a distance limit, if any distance is found to be below this limit, no further pairs are tried (default TINY(1.0D0)).
To use this keyword a `metric' file is required. The `metric' file takes the following format, with one line entry for each minimum in the database:
metric1 nmin [metric2] [metric3], metric1 takes priority over metric 2 etc...
You have to construct this file manually with your own choice of metric(s).
You also need a `metric.ordered' file - this speeds up the finding of pairs.
In /SCRIPTS/PATHSAMPLE/ there is a program to convert the metric file and produce a metric.ordered file, `getmetricordered.f90'.
However, you won't normally want all minima in the `metric.ordered' file. Ideally, this is a list of minima in the main basin to which connection attempts should be made, usually the main basin can be defined by an high energy cutoff.
Running disconnectionDPS with the keyword BASINT followed by a particular energy gives the results of a basin analysis at this energy in a file `basins'.
`getmetricordered.f90' will produce a `metric.ordered' file from `metric' and `basins', if you set the required input arguments.
The metric and metric.ordered files must be constructed manually and are currently not updated during a run when minima are added to the database.
Different metrics are required for different systems.
- USEPAIRS Epathfile: pairs of minima are chosen for connection attempts based
on the order of local minima specified in file Epathfile, which must have the format
of a PATHSAMPLE Epath output file 9as generated by DIJKSTRA, etc.
This keyword is intended to follow a DUMMYTS run, where the stationary point
database has been refined using BHINTERP or BISECT runs in OPTIM.
The dummy ts.data and pairs.data files must be renamed or removed
before the USEPAIRS run.
The pairs of minima are first chosen to be the adjacent structures from the list, then
second-neighbours, until all possible pairs have been tried.
This keyword can also be used to specify a list of pairs to attempt to connect. See the MAKEPAIRS keyword to help assemble Epathfile in this case.
![\includegraphics[width=0.5\textwidth]{PHE.eps}](img37.png)