Input is keyword driven with sensible defaults in most cases.
Free format may be used within each line. Blank lines are ignored.
- 2D: enforce two-dimensional `flatland'.
- A9INTE: specifies that after each quench that does not lead to an inversion of chirality,
isomerisation of a peptide bond or cold fusion - the interaction enthalpy between a specified residue and the rest of the system
should be calculated using the external script `AMBGMINintE.sh', and read back into GMIN. This is intended for
use with protein/ligand systems where you are searching for low energy docked structures. As the total energy
does not fully correlate with the protein/ligand interaction enthalpy, it is often useful to retain not only the
lowest SAVE total energy structures, but also the lowest SAVEINTE interaction enthalpy structures.
To use this keyword, the `AMBGMINintE.sh' script (contained in the SVN repository in the SCRIPTS directory) must
be present in the GMIN working directory. You should ensure that you have edited it to match the residue numbering
of your system. You also need a full AMBER9+ installation with access to the `sander' executable. When using this
keyword, an interaction enthalpy dump file is produced every DUMPINT steps, and at the end of the run,
structural output files are produced for the SAVEINTE lowest interaction enthalpy geometries. After each quench,
the structure with the current lowest interaction energy is dumped in pdb and rst format prefixed with `bestint.' to allow
monitoring.
- ACCEPTRATIO accrat: accrat is the required acceptance ratio for the MC
exploration of the transformed surface. For fixed temperature runs (the default) the maximum step size
is adjusted to try and meet the requested value of accrat; for a fixed maximum
step size the temperature is adjusted instead. The default value of accrat is a half.
- ACE step: used for regular Born radii updates in CHARMM every step steps.
- ACKLAND id: specifies an Ackland embedded atom metal potentialcoded by Dr Mihai-Cosmin Marinica.
id specifies the particular metal: 1 is ?, 2 is ?, 3 is ?, 4 is ?, 5 is iron, 6 is a different iron,
7 is tungsten.
Positive values for id specify periodic boundary conditions, where box lengths must be
specified by the PERIODIC keyword.
Negative values for id specify a cluster calculation. A CUTOFF value can also
be used for clusters.
- ACKLAND1: specifies first iron potential.
- ACKLAND2: specifies second iron potential.
- ALGLUE: specifies a glue potential for aluminium.
- ALIGN: specifies that, before the basin-hopping run, a second set of coordinates should be read in from finish, and an alignment operation between the coordinates in coords and the set in finish is performed. This is very similar to the keywords PERMOPT and PERMINVOPT, but unlike those it doesn't require that permutations be specified. Because of the way those keywords are set up, the minimised distance between the two structures is returned as an ''energy'' and therefore will cause an error when the basin-hopping run starts. For this reason, ALIGN should not be used with non-zero STEPS. This keyword should only be used for testing alignment algorithms. OPTIM is the appropriate tool for aligning structures.
- AMBER12: specifies calculation with the interfaced version of the AMBER 12
pmemd program. AMBER 12 requires an additional input file, min.in, which specifies
keywords for the AMBER 12 potential. It also requires appropriate topology and coordinates, in files
named coords.prmtop and coords.inpcrd respectively. For details, see the AMBER 12 manual.
As with the AMBER 9 interface, cutoffs are smoothed, using the mini̇n keyword ifswitch=1.
Additional keywords are as AMBER9, with the exception of AMBERMDSTEPS, which is not implemented.
- AMBER20 start inpcrdformat: specifies calculation with the interfaced version of the AMBER 12
pmemd program.
Starting coordinates are read from start (default coords.inpcrd), in Amber inpcrd
file format specified by the second optional argument inpcrdformat.
If the second argument is missing, it is assumed that start contains
only three columns with the xyz coordinates of all atoms, in the same order
as in the topology file.
AMBER 20 requires an additional input file, min.in, which specifies
keywords for the AMBER 12 potential. It also requires appropriate topology and coordinates, in files
named coords.prmtop and coords.inpcrd respectively. For details, see the AMBER 20 manual.
As with the AMBER 9 interface, cutoffs are smoothed, using the mini̇n keyword ifswitch=1.
Additional keywords are as AMBER9, with the exception of AMBERMDSTEPS, which is not implemented.
- AMBER9 start inpcrdformat: specifies a calculation with the interfaced
version of the Amber 9 program package. From this package the Amber force fields
are being used, with small modifications (e.g. smooth cut-offs).
Starting coordinates are read from start (default coords.inpcrd), in Amber inpcrd
file format specified by the second optional argument inpcrdformat.
If the second argument is missing, it is assumed that start contains
only three columns with the xyz coordinates of all atoms, in the same order
as in the topology file. To start a run with this interface,
several auxiliary files are required in the same directory: input coordinate file
coords.inpcrd, parameter topology file coords.prmtop,
input file to Amber containing force field specifications min.in, and, if
desired, a coordinate file different from coords.inpcrd containing
starting coordinates.
To turn on smooth cutoffs for the Generalised Born force fields, the keyword
ifswitch=1 has to be used in the &cntrl namelist block of min.in.
When using the AMBER9 keyword, any calculated second derivatives will be
numerical. If one wants analytical second derivatives, the NAB keyword
should be used instead, with the same syntax.
Additional keywords for the AMBER 9 runs are DUMPSTRUCTURES, AMBERMDSTEPS,
LIGMOVE, BLOCKMOVE and MOVABLEATOMS.
- AMBERENERGIES dump energy components for AMBER at every step.
- AMBERMDSTEPS MD steps for AMBER9 as propagation steps in basin-hopping. Rather inefficient method.
- AMBERMUTATION nmut ntest Mutational steps coupled with group rotations for AMBER12.nmut is the frequency of mutational steps attempted, and ntest the number of steps after the mutation before a Metropolis criterion is applied to accept or reject the mutation.
- AMBERMUTENERGY opt1 opt2 Specifies what energy or penalty function should be used to accept or reject mutations. opt1 can be 1 (random number, for testing), 2 (a component of the AMBER energy, e.g. solvation energy; opt2 specifying which term), 3 (interaction energy between two molecules, opt2 is the last residue of the first molecule), and 4 to 7 for various geometrical measures for helices.
- AMBERMUTFF ff The used force field to recreate the topology files as needed, ff is currently implemented for 99 (ff99SB) and 14 (ff14SB).
- AMBERMUTIGB igb Solvent model used. igb is either 2 or 8.
- AMBERMUTRIGID
- AMCHNMAX: The maximum number of angles that will be changed by up to STEP during an
AMBER dihedral step. If this is not set or is set to zero, cartesian steps of maximum size STEP are taken
instead.
- AMCHNMIN: The minimum number of angles that will be changed during an AMBER dihedral step.
- AMCHPMAX: The maximum probability for a single angle to be twisted in an AMBER dihedral step.
- AMCHPMIN: The minimum probability for a single angle to be twisted in an AMBER dihedral step.
- AMH Associative Memory Hamiltonian protein model.
- ANGSTROM: specifies coordinates in Ångstrom for the FRAUSI
potential.
- ANSATZ: specifies that ANSATZGMIN is running instead of regular GMIN for combined QC1 and QCUCC calculations.
To compile, set WITH_ANSATZ and WITH_OPENMP to ON in INTERFACES. If working with complex numbers, optionally set WITH_ANSATZCOMPLEX to ON.
- ARGON: introduces a diatomics-in-molecules calculation for
a neutral, cationic or electronically excited argon cluster. See also
GROUND, PLUS, TWOPLUS and STAR.
- ARM arma armb: use the acceptance-ratio method (Bouzida et al., Phys. Rev. A,
45, 8894, 1992) to adjust the step size to achieve the requested
acceptance ratio. A scaling factor is calculated and applied to step, rotmax,
and/or transmax. The scaling factor is calculated according to
log(arma*Pt + armb)/log(arma*P0 + armb), where Pt defines the
target acceptance ratio and P0 the actual acceptance ratio. Both values arma and
armb default to 0.4.
- ARNO: specifies a diatomics-in-molecules potential for ArN-NO clusters.
- ATOMMATCHDIST: used for bulk systems, providing an alternative method for finding the shortest distance between structures.
This method is particularly useful for finding the shortest distance between very similar structures such as defective crystal structures.
The method is an extension of the original bulkmindist.f90 routine for matching permutationl isomers. Atoms are overlayed and the number of exactly matching
g atoms is maximised. (ATOMMATCHINIT uses atom matching only for aligning initial structures and then reverts to the default distance method.)
With ATOMMATCHDIST/INIT, the method is non-deterministic to maximise efficiency. It has been optimised to work particularly well for providing small distan
ces between similar structures and for removing very large distances. However, it will not always outperform the default method for middling distances.
Currently only one method for calculating distances, atom matching or the default, can be used.
- ATOMMATCHFULL: As for ATOMMATCHDIST, but provides a slow deterministic result that should always outperform or equal the default distance calculation and ATOMMATCHDIST but may never finish.
- AUCMLPLOG: Logical, if included, a log function is used as loss function instead of AUC.
This keyword makes the AUCMLPVB3 routine equivalent to the original MLPVB3 routine.
Default is FALSE.
- AUCMLPVB3 in hidden out data λ β: Specifies a single-layered neural network as with keyword MLPVB3 (see below),
with in input nodes, hidden hidden nodes, out output nodes and λ as the L2 regularisation parameter.
data is the number of data points the model trains with, stored in a file called MLPData. This file should be
formatted as follows:
| y1 |
x11 |
x21 |
x31 |
... |
| y2 |
x12 |
x22 |
x32 |
... |
| y3 |
x13 |
x23 |
x33 |
... |
| ... |
|
|
|
|
|
yNdata |
x1Ndata |
x2Ndata |
x3Ndata |
... |
where yi is the outcome for data point i, and xji is the jth input
feature for data point i. The features will be read starting from the
feature in column start (ignoring the y column).
Unlike MLPVB3, instead of calculating the associated loss value, an approximated AUC is calculated.
The true AUC value is given by the following non-differentiable function:
AUC(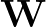 ; ;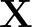 ) = ) = 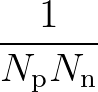   Θ Θ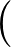 p1(; p1(;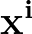 ) - p1(; ) - p1(;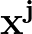 ) ) , , |
(1) |
where Θ(x) is the Heaviside step function (1 for x > 0 and 0 otherwise). This function is not differentiable, so we must use an approximate function.
The function to be minimised is
p1( ;
;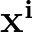 ) is the softmax probability of the neural network giving output class 1 for data point i, given weights .
The sums are over data points where the true class is 1, of which there are Np, and over data points where the true class is not 1, of which there are Nn.
β is the parameter for the sigmoid function
) is the softmax probability of the neural network giving output class 1 for data point i, given weights .
The sums are over data points where the true class is 1, of which there are Np, and over data points where the true class is not 1, of which there are Nn.
β is the parameter for the sigmoid function
σ(z) 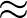 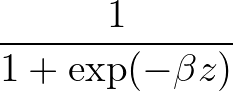 . . |
(3) |
Larger values of β make curve steeper, hence approximate the true AUC more closely.
The maximum value allowed for β is set at 500 to prevent floating-point underflow errors.
If β is set higher than this value, the maximum value of 500 will override it and be used instead.
- AVOID dist maxsave: specifies that the geometry should be reseeded if the
latest structure gets within a distance dist of the maxsave members of a
cyclic list. If a third argument `F' is added to the AVOID line then such
steps are simply rejected rather than reseeded.
The NEWRESTART keyword must be specified to populate the list of
minima to be avoided. If the `F' argument appears for AVOID then
NEWRESTART will not reseed.
- AXTELL zstar: specifies an additive Axilrod-Teller term for certain
diatomics-in-molecules potentials as well as the Pacheco-Ramelho intermolecular potential for
C60.[#!pachecor97!#]
zstar is the coefficient multiplying this term.
- BASIN bgmax: specifies a basin-hopping run (as opposed to standard MC
on the untransformed surface). bgmax is the convergence threshold
on the RMS force in the basin-hopping
quenches. If this criterion is too strict then the run time will be greatly increased.
If it is too sloppy then the performance of the algorithm is impaired. Different values
are needed for different potentials. SLOPPYCONV can be used instead.
- BENZRIGIDEWALD ewaldrealc ewaldrecipc: calls anisotropic potential developed by [#!TottonMK10!#]
for periodic systems of rigid benzene molecules and uses Ewald summation to compute the long-range electrostatics.
ewaldrealc and ewaldrecipc specify the real-space and reciprocal-space cutoff values for
the Ewald summation. The cell dimensions should be specified using the PERIODIC keyword. This potential
works for both orthorhombic and triclinic cells. Also, the RIGIDINIT keyword must be specified. Also, the
energy gradient with respect to cell parameters is computed if the BOXDERIV keyword is used.
- BFGS: specifies that the full BFGS minimiser should be used. Inefficient compared to LBFGS.
- BGUPTAT NTYPEA AAA PAA QAA ZAA R0AA: One of the required keywords to specify a Binary Gupta run.
NTYPEA is specified, followed by the potential parameters for the A-A interactions. See also BGUPTATAB and BGUPTATBB.
[NB: Per-atom energies will be stored.]
- BGUPTATAB AAB PAB QAB ZAB R0AB: The line to specify the potential parameters for the A-B interactions
for a Binary Gupta run.
- BGUPTATBB ABB PBB QBB ZBB R0BB: Specifies the B-B interaction parameters.
- BGUPTANAME name1 name2: specifies the names of species A and B for a Binary Gupta run. Default are Au and Ag.
- BHPT pttmin pttmax exchprob (RANDOM|INTERVAL) (SINGLE|SETS) seed:
specifies a parallel tempering basin-hopping run with temperatures exponentially distributed between pttmin and
pttmax. Either the probability of attempting replica exchange (float,
0.≤x < 1.) or the corresponding mean interval
in terms of basin-hopping steps
(integer, x≥1) may be supplied as the parameter exchprob. These exchanges may be attempted at random or at intervals (default: RANDOM,
alternative is INTERVAL).
At each exchange event, either a single exchange between a random pair adjacent replicas is attempted, or exchange between all
pairs in either the set of even pairs,
{{1, 2},{3, 4}...} or set of odd pairs,
{{0, 1},{2, 3}...} (default: SINGLE,
alternative is SETS).
To specify SINGLE or SETS the RANDOM or INTERVAL parameter must appear first.
seed is an integer that will be used as the random number seed.
BHPT should be used together with the MPI keyword to specify an MPI parallel job.
The number of replicas corresponds to the number of MPI tasks, which is
defined in the sbatch script. For example, for 20 tasks:
#SBATCH -n20 -N1
#SBATCH –tasks-per-node=20
# and then the job itself
mpirun -n 20 /sharedscratch/wales/GMIN.gfortran.mpi/GMIN > output
The source must be compiled with MPI enabled.
If DUMPSTRUCTURES is specified, then every DUMPINT steps, minima
from each replica will the written out in XYZ format to files dumpmin.replicaNumber.minimaNumber.
For potentials that read the coordinates from coords the replicas will be seeded from that file with
configurations read in replica order. Hence the number of coordinate entries in coords must equal the
number of MPI replicas. For AMBER12 and AMBER20 coordinate entries can be read for the different
replicas if a corresponding start file is provided and specified by AMBER20 start or
AMBER20 start. The number of entries in the start file must be the same as the number of MPI replicas.
If AMBER input format is used via an inpcrd file then all the replicas will be seeded with
the same coordinates from that file.
BHPT is currently not compatible with periodic variable cell optimisations via BOXDERIV. The BOX_PARAMS
variables have not been parallelised yet.
- BLOCKMOVE nblocks block_1 block_2 ... block_n: used with AMBER9, MOVABLEATOMS and LIGMOVE.
Divides the atom list in the `movableatoms' file into nblocks distinct blocks that are treated independently as
rigid during steptaking moves. block_i specifies the number of atoms in each block. Step size parameters for rigid
rotation, translation and cartesian moves are taken from the parameters of LIGMOVE.
- BINARY ntypea epsab epsbb sigmaab sigmabb: specifies a binary Lennard-Jones
system. ntypea is the number of type
A atoms—the rest are assumed to be type B and appear at the end of the list
of coordinates.
εAA = σAA = 1 define the units of energy and length,
and epsab=
εAB, epsbb=
εBB,
sigmaab=
σAB, sigmabb=
σBB.
The box parameters and cutoff should be specified with the PERIODIC keyword.
- BINARY_EXAB freq: attempt exchange move for an A and a B particle every freq steps.
- BINSTRUCTURES SaveNth: requests that the geometry of every SaveNth
new structure found during basin-sampling is
recorded in binstructures.j, where j is the index of the bin
to which a given minimum belongs. If this keyword is
present then GMIN switches from plain PTMC to BSPT.
Without BINSTRUCTURES the BSPT keyword will perform a
standard PTMC run with no quenching.
- BLJCLUSTER ntypea epsab epsbb sigmaab sigmabb cutoff: specifies a binary Lennard-Jones
cluster. The parameters are the same as for BINARY, above.
- BLJCLUSTER_NOCUT ntypea epsab epsbb sigmaab sigmabb: specifies a binary Lennard-Jones
cluster without a distance cutoff. More efficient than BLJCLUSTER for smaller systems.
The parameters are the same as for BINARY, above.
[NB: Per-atom energies will be stored.]
- BLN kr kθ : specifies a BLN off-lattice protein model with
bond-length and bond-angle force constants kr and kθ.
An auxiliary file BLNsequence is required.
See §
![[*]](crossref.png) for more details.
for more details.
- BLNGO kr kθ λ: specifies a Gō potential
with the same form as the BLN potential. The parameters kr
and kθ are the same as those used for the BLN keword and a BLNsequence file is required. Also needed is an auxiliary file, contactmap, containing the pairs of residues in contact in the native state of the protein
with one pair of residue numbers in each line of the file. An optional
parameter, λ, specifies the strength of the non-native interactions in a
scaled BLN potential [#!KimKS09!#].
- BOXCENTROID x y z dx dy dz (ix iy iz): confine the system centroid to region (x±dx, y±dy, z±dz). Intended for systems interacting with an external field, e.g. a cluster supported on a substrate modelled using keyword MIE_FIELD. The cluster centroid will be checked after each random perturbation (before quenching!), and on each step of the QALCS_SURF procedure (if used, and also before quenching). When a centroid coordinate (say x_c) ventures outside the corresponding range (x±dx), that centroid coordinate is translated back to x. If any of the optional integer parameters (ix, iy, iz) are set to unity (default value is zero), then the translation vector(s) in the corresponding direction(s) will be restricted to integer multiples of dx and/or dy and/or dz. This additional feature is intended for periodic external fields, e.g. a periodic substrate, in which case dx/dy/dz ought to match the period.
- BOXDERIV boxlx boxly boxlz alpha beta gamma: specifies optimisation of the cell parameters for periodic systems.
No argument is needed if the cell parameters are otherwise known
e.g. read from dftb_in.hsd in a DFTBP setup, or from the system.cell file for CASTEP,
or the POSCAR file for VASP.
For CASTEP and VASP the BOXDERIV directive also sets USEFRAC so that the atomic coordinates are
manipulated in fractional form during the optimisation.
For MANYBODY SW the coordinates are Cartesians and read from the coords file; they are not fractional.
The angles on the BOXDERIV line should be in degrees.
Otherwise, boxlx, boxly, boxlz, alpha, beta, gamma are the
box lengths and angles, in radians, of the initial structure. If only the box lengths are given, the cell will be taken as
orthorhombic, and only the cell lengths will be optimized. The cell lengths and angles of the initial structure should also
be specified using the PERIODIC keyword. If the gradients wrt cell parameters are being calculated for a system
using atomic, Cartesian coordinates, the initial coordinates in the coords file should be in fractional coordinates
for VASP and CASTEP. If
the gradients wrt cell parameters are being calculated for a system using rigid body coordinates, the initial coordinates
coordinates should be given in the usual way for GENRIGID, and the RIGIDINIT keyword should be used.
- BOXSTEP boxstepfreq: for use with the framework for optimizing cell parameters using the BOXDERIV keyword.
Randomly changes the cell lengths and angles every boxstepfreq basin-hopping stes.
- BOXPRESSURE pressure: for use with variable cells and the BOXDERIV keyword. Uniformly applies pressure GPa to the cell via the stress tensor. A corresponding p*V term is added to the energy, making it the enthalpy.
- BRANCHNBOUND N: Specify the use of the GO-PERMDIST alignment algorithm described in Griffiths et al., JCTC 2017. This algorithm overrides the default MINPERMDIST alignment subroutine. GO-PERMDIST uses a branch and bound algorithm combined with the older PERMDIST method for combined translation-rotation alignments. It can be used for either periodic or aperiodic systems. Use of GO-PERMDIST is recommended for aperiodic systems, but for periodic it can be very slow, and use of the FASTOVERLAP keyword is recommended instead.
GO-PERMDIST is deterministic and, if allowed to run indefinitely, will guarantee identification of the best alignment. In practise it is run with an early exit criterion expressed as a number of iterations N. A small value of N is often sufficient to identify the best possible alignment but usually insufficient to prove that the identified alignment is the best possible value. The default is 2000, which is generally a reasonable compromise between speed and accuracy.
- BSMIN: specifies a Bulirsch-Stoer minimisation scheme.
Very inefficient compared to LBFGS.
- BSPT histmin histmax ptemin ptemax pttmin pttmax exchprob nequil ptsteps nquench nenrper hbins qfrq:
requests a basin-sampling run to accumulate the quench probability for local minima
as a function of potential energy using
a parallel-tempering algorithm.
This keyword also specifies the energy range for the histogram of quench energies,
histmin to histmax,
the energy range for the histogram of instantaneous configurations, ptemin to ptemax,
the temperature range (pttmin and pttmax),
the probability of attempting an exchange exchprob, the
number of equilibration steps, nequil,
the number of parallel tempering MC steps without quenching, ptsteps,
the number of parallel tempering MC steps with quenching, nquench,
the number of bins for the histogram of instantaneous potential energy, nenrper,
the number of bins for the histogram of quench energies, hbins,
and the quench frequency, qfrq.
Should be used together with the MPI keyword. (This option is only available if the source is compiled with an MPI enabled.)
- BSPTDUMPFRQ n, n is the interval at which intermediate statistics
and bsptrestart files are dumped. If n is less than one these files
will only be dumped at the end of a complete run.
See also BSPTRESTART.
- BSPTRESTART: restart a previous BSPT or PTMC run.
The instantaneous and quench potential energy histograms are read from the last
Visits.his and Visits2.his files, and the current state from
bsptrestart files (one per node, numbered from zero).
A finished run can be continued with more steps by changing the nquench
or ptsteps parameters on the BSPT or PTMC line of
the data file. Setting the interval for BSPTDUMPFRQ to
minus one will read the last set of dump files.
- BSPTQRANGE minE maxE: set minimum (minE) and maximum (maxE) energies for BSPT quenches.
- CALCQ: turn on calculation of bond order parameters.
- CAMSHIFT csversion svnroot shiftfile csn csalpha: uses chemical shifts as restraints during the
optimization procedure. Currently, CAMSHIFT can only be used together with CHARMM.
csversion is a string that specifies the method for combining the two potentials: MERGE means
(1 - α)*Charmm + α*Camshift,
ORIGINAL means
Charmm + α*Camshift, and NOFF means
α*Camshift.
svnroot specifies the svn root directory (e.g., $HOME/svn/). shiftfile is the file containing the
experimental shifts, which has to be located in the working directory. csversion, svnroot and shiftfile all have to be
specified together with CAMSHIFT. csn and csalpha are optional parameters. They define the
tolerance parameter of the CamShift energy profile, and the relation between CamShift and the force field, respectively.
Default values for both are 1.0.
- CAPSID rho epsilon radius height: specifies a coarse-grained potential to represent virus capsid pentamers
with parameters ρ,
ε2, r and height, respectively.
If height is omitted the default is 0.5.
- CASTEP job system: tells the program to use the specified CASTEP
executable to calculate energies and forces with input files based on the system name system.
job is a string in quotes that specifies the system call required to run the CASTEP executable.
OPTIM will add a line with `elec_restore_file' to the param file after the first call to obtain the CASTEP energy and gradient.
This saves a great deal of cpu time in subsequent CASTEP steps.
However, it means that the final param file is altered, and you will need
to remove the elec_restore_file line from the param file to restart
the job if valid wavefunction files are not present in the working directory.
CASTEP will fail and stop if the wavefunction read fails.
Note that you cannot restart a job with a different number of nodes using saved wavefunctions!
It is also necessary to specify the atoms in order of increasing atomic number,
since otherwise they are reordered by CASTEP. There is now a check for this in OPTIM.
The system.cell file is saved in system.cell.old in each call to potential before CASTEP is run.
The geometry at the start of each Rayleigh-Ritz optimisation is saved in system.cell.save.
To restart a transition state search the system.cell.save should generally be used.
The PARALLEL keyword is no longer needed, since the number of processors can be specified in job if necessary:
CASTEPC 'srun -N 1 -n 10 castep.mpi' 'qge'
For an interactive run on nest mpirun works:
CASTEPC 'mpirun -np 10 castep.mpi' 'qge'
For a serial run on nest:
CASTEPC 'castep.serial' 'qge'
sbatch runs on nest require:
module add gcc/7.5.0
module add mpi/openmpi/gnu7/4.0.5
module add castep/19.1/intel17-openmpi-gnu7
See also GUIDECASTEP for guiding with xtb.
- CENTRE : if present the system will be translated so that the centre-of-mass
lies at the origin after every quench.
- CENTREXY : if present the system will be translated so that the centre-of-mass
lies at the centre of the xy plane after every quench. This is useful when using an implicit membrane like IMM1 where you have directionality only in the
z-direction, so centreing in x and y should have no delaterious effect.
- CG : specifies a conjugate-gradient minimisation scheme. Inefficient compared to LBFGS.
- CHANGEACCEPT naccept: naccept is an integer which sets the interval
at which the acceptance ratio is checked and possible adjustments are made to the maximum
step size or the temperature. The default is naccept= 50.
- CHARMM: specifies that a CHARMM potential should be used.
See also keywords CHARMMTYPE, CHPMAX, CHPMIN, CHNMAX, CHNMIN,
NOPHIPSI, TOMEGA, INTMIN, CHFREQ, CHRIGIDROT,
CHRIGIDTRANS, and RMS. If CHNMAX is not specified, a cartesian
displacement step taking scheme will be used. For cartesian steps, rings are moved as rigid bodies to avoid false knotted minima. See RINGROTSCALE. Finally, Molecular Dynamics (MD) can be employed to generate new geometries. See CHMD
- CHARMMDFTB: specifies that the CHARMM SCC-DFTB potential is to be used, and
disables updates of the nonbonded list. This assumes you are using a fully QM system. If you
are using a QM/MM setup, you should not use this keyword! Note that SCC-DFTB can only be used
with CHARMM35.
- CHMD CHMDFREQ: Requests Molecular Dynamics (MD) runs to be performed every CHMDFREQ step to generate new geometries. A CHMDFREQ setting of 20 will execute an MD run every 20
th step, while dihedral or cartesian moves are applied otherwise as specified in the data file. A CHARMM parameter file named 'chmd.par' containing all relevant keywords for the CHARMM DYNA module has to be present in the working directory. All CHARMM keywords must be uppercase and given in the first line. A typical example is:
VERL NSTEP 500 TIMESTEP 0.002 TWINDH 10.0 IEQFRQ 200 ICHECW 1 IASORS 0 IASVEL 1 FIRS 500 FINA 500
Please consult the CHARMM manual for further details on the DYNA module. Currently, the length of the input string given in 'chmd.par' is limited to 500 characters.
- CHARMMENERGIES: prints the components of the total CHARMM energy after each step.
- CHARMMTYPE topfile paramfile: topfile and paramfile are the
common CHARMM top and param files , e.g., `toph19_eef1_perm.inp' and `param19_eef1_perm.inp'.
- CHFREQ nfreq: used with CHARMM keyword to specify that every
nfreq basin-hopping steps dihedrals are twisted. Default is nfreq=1.
- CHNMAX: used with CHARMM keyword to specify the maximum allowed
number of angles to be twisted. Specifies a dihedral angle step taking scheme.
- CHNMIN: used with CHARMM keyword to specify the minimum allowed
number of angles to be twisted.
- CHPMAX: used with CHARMM keyword to specify the maximum allowed
probability for twisting an angle.
- CHPMIN: used with CHARMM keyword to specify the minimum allowed
probability for twisting an angle.
- CHRIGIDROT prot rotmax nrot: used with CHARMM keyword
to support rigid body rotation every nrot basin-hopping steps with maximum allowed
probability prot and maximum allowed rotation angle rotmax (in degrees).
The keyword CHRIGIDROT requires a file segments.tomove, which specifies
the segments for rigid rotation. The segments are numbered and each line contains only one number.
- CHRIGIDTRANS ptrans transmax ntrans: used with CHARMM keyword
to support rigid body translation every ntrans basin-hopping steps with maximum allowed
probability ptrans and maximum allowed translation transmax (in Å).
The keyword CHRIGIDTRANS requires a file segments.tomove, which specifies
the segments for rigid translation. The segments are numbered and each line contains only one number.
CHRIGIDROT and CHRIGIDTRANS use the same segments.tomove.
- CHECKD n: calculates gradients analytically and numerically for the initial coordinates, then exits. n is an
integer optional argument; if equal to zero, then only the single-point energy is calculated and reported.
- CISTRANS: disables all checks for cis or deformed amide/peptide bonds.
- CHIRO σ0 μ γ [L]: calls the OK potential. If
L is absent or 0, then use one LJ site. If L > 0, use LJ-like rods of
length 2L. Output is written to chiro.xyz. A periodic boundary
condition in the x direction can be applied using the PERIODIC keyword.
- CNBH: a critical nucleus basin-hopping run. As for FEBH (must also be set)
but the accept/reject move for sizes is based on the minimum partition function (maximum free energy)
for competing sizes. This is the superposition sum for all the known distinct minima of the
corresponding sizes.
- COLDFUSION thresh: if the energy falls below threshold thresh then
cold fusion is assumed to have occurred and geometry optimisation stops.
The default value is -106.
- COMPRESS kcomp kpower: add a compression (or expansion) potential with force constant kcomp
and power kpower using the centre-of-coordinates distance for each atom.
The compression term can be turned off once a given RMS tolerance is achieved
using the GUIDE keyword.
This combination can be used with the generalised rigid body scheme introduced via the
RIGIDINIT keyword.
- COMPRESSO kcomp: add a harmonic compression potential with force constant kcomp using the
distance from the origin for each atom.
- COMPRESSRIGID kcomp dist: if at least one rigid body's centre of mass is further than dist from its nearest neighbour,
add a harmonic compression potential with force constant kcomp using the centre-of-mass distance for each rigid body. The compression is
disabled once the rigid bodies are all within dist of another.
This keyword is only implemented for a few specific potentials.
However, the COMPRESS and GUIDE keywords can be used with the general rigid body scheme
introcduced via GENRIGID.
- COMMENT : the rest of the line is ignored, as for !. A space is required before any characters on the
rest of the line.
- CONVERGEDONLY : structures are only accepted into the saved list and the Markov chain if
the minimisation has converged. For potentials with unphysical regions and discontinuities it is possible
for a minimisation to fail at an energy below the global minimum but above the COLDFUSIONLIMIT,
and we need to reject these structures. For well behaved potentials we might want to accept structures
with low energies where a minimisation runs out of steps. The default behaviour is to accept configurations
with low enough energy even when a minimisation has not converged.
- COOPMOVE n cut: specifies cooperative moves in the step-taking routine. An atom is
selected at random, and the n nearest neighbours (default 5) that lie within a cutoff
distance of cut (default 1.0) are moved by the same amount.
- CPMD sys: specifies that the CPMD program should be called for energies and gradients. Not
tested!
- CSM pg opt2 opt3 : continuous symmetry measure from Pinsky, Dryzun, Casanova, Alemany and Avnir, J. Comp. Chem., 29, 2712, 2008. Point group pg needs to be specified.
- CUDA potential: specifies a GPU run, using the CUDA C++ implementation. Do not use for the CUDA Fortran implementation. Setting potential to 'A' specifies the AMBER12 potential. See the group wiki for further information.
- CUDAFORLBFGS: specifies that the CUDA Fortran implementation of LBGFS be used, only available when the executable was compiled with
CUDA Fortran. This option is likely to signifcantly slow down minimisation unless there are tens of thouands of degrees of freedom. A small number of
UPDATES is recommended when using this option, typically 4.
- CUDATIME: get timings for CUDA runs (benchmarking).
- CUTOFF cutoff: sets a cutoff beyond which the potential is truncated. This
only has an effect for tight-binding silicon at present. Interaction cutoffs for other potentials
should be specified in their input files e.g. min.in (and min_md.in if used)
for AMBER9 or below the CHARMM line for CHARMM.
- DAMPEDGROUPMOVES: damped group moves, the structure is very similar to group rotation moves with appropriate changes.
- DBP epsbb sigmabb muD E: calls
a finite system of dipolar Lennard-Jones dumbbells [#!ChakrabartiFW09!#], where the electric field of stength E can be
optionally present. The field, if present, is along the space-fixed z-direction.
εaa
and
σaa are both set to unity by default. μD is the dipole moment; ε's
and σ's correspond to the Lennard-Jones parameters.
- DBRENT: specifies minimisation using Brent's method with first derivatives in the
conjugate-gradient procedure.
Inefficient compared to LBFGS.
- DEBUG: sets various debug printing options including the dumping of initial
geometries and energies (to dump.X.xyz) if DUMP is also set.
- DECAY x: magnitude of random move decays according to parameter
x with distance from a randomly chosen atom.
- DF1: specifies a binary 2D potential.
The first N/2 atoms have unit radius and the rest
have radius 1.4, with a cutoff for each pair type at the
average radius.
The keyword 2D must also be specified, along with a
PERIODIC line to specify two box-lengths.
Initial work uses box lengths of 3.31437171 for a number density of 0.9.
- DFTB: specifies a DFT-based tight-binding potential; the multiplicity is specified by
keyword MULTIPLICITY.
- DFTBP: specifies that the DFTBP (SCC-DFTB+) code is used. The required files are dftb_in.hsd and the relevant Slater-Koster
parameter files (*.skf) for each atomic pair in the system under study.
These SK files can be downloaded from: https://dftb.org/parameters/download/. SK files are not required if a GFN1-xTB or GFN2-xTB Hamiltonian is used via DFTBP.
The geometry should always be specified in the dftb_in.hsd file; a start file is ignored.
How to build DFTBPGMIN
To build DFTBPGMIN either use Intel and an mkl library, or GCC. Other modules may interfere. The following examples were configured on nest:
- Configure the DFTBP submodule source in ~/softwarewales/ by executing
git submodule init DFTBP
git submodule update
- Configure the build with either Intel or GCC:
| a. | For an Intel build load only the appropirate modules:
cmake/3.23.2 idb/latest icc/64/2022/0/1 tbb/64/2022/0/1 oneapi-compiler-rt/64/2022/0/1 mkl/64/2022/0/0
Always specify the compiler environment variables when configuring the build
FC=ifort CC=icc CXX=icc cmake ~/softwarewales/GMIN/source/ -DWITH_DFTBP=yes
and then
make
|
| b. | If using GCC, a system BLAS needs to be used. Depending on the machine, CMake may be able to find one, or you may have to
specify one yourself. This combination of modules works on nest:
gcc/12.2.0, cmake/3.23.2
with a configure line of
cmake ~/softwarewales/GMIN/source/ -DWITH_DFTBP=yes -DWITH_MYBLAS=no
-DBLAS_LIBRARIES=/usr/lib64/libopenblas.so.0
|
- (optional) With the latest git submodule configuration a standalone dftpb binary should automatically be built when compiling DFTBPGMIN or DFTBPOPTIM. Manually, the binary can be created while in the DFTBP directory by using
FC=ifort CC=icc cmake -DCMAKE_INSTALL_PREFIX=$HOME/softwarewales/DFTBP/_build -B _build .
then
cmake --build $HOME/softwarewales/DFTBP/_build -- -j
then
cmake --install $HOME/softwarewales/DFTBP/_build
- DFTBPBIN binary: Specify a dftb+ binary to use. This will switch the program from the DFTBP API into a subprocess version and must be used together with the DFTBP keyword! Since dftb+ will be executed as systemcall, no dedicated DFTBPGMIN binary is required when using DFTBPBIN.
- DFTBPRESET: Tell DFTBPGMIN to re-initialize the DFTBP API at every potential call. This might be necessary to avoid a drift of the energy and is done by default.
- DFTBPNORESET or NODFTBPRESET: Tell DFTBPGMIN to NOT re-initialize the DFTBP API at every potential call.
- DFTBC: specifies a DFT-based tight-binding potential carbon.
Can be guided using LJAT.
- DGUESS dguess: initial guess for diagonal elements of the inverse
Hessian, used whenever the LBFGS optimiser is reset.
The default is dguess=0.1.
- DIHEDRALROTATION: This keyword performes random rotations of groups about specified dihedral angles. Similar to GROUPROTATION, the dihedral groups must be defined in a file named dihedralgroups, with the following syntax:
GROUP name type at1 at2 at3 at4 N maxrot pselect
gat1
gat2
…
gatN
Here ``name'' is a string defining the group name; ``type" is a string defining the type of dihedral for backbone rotations (phi, psi etc.), used for printing purposes only; at1 to at4 are integers specifying the atom indices defining the dihedral angle, gat1… gatN define indices of the atoms which rotate, and N the number of such atoms.
Note: at4 should also be included in the list of atoms to rotate, as it is the final atom defining the dihedral!
- DIPOLES: causes the first order induction energy to be included
in a diatomics-in-molecules calculation for Ne+n or Ar+n. By default this
term is neglected, although it may be significant.
- DISTCON file: specifies a general atom-atom harmonic distance constraint potential, which is added
to the underlying potential. file is a file containing a list of atom pairs, the reference distance, and the
force constant used in the harmonic term that penalises deviation from the reference separation. For example
367 393 2.27 1.01D3
372 385 2.65 1.02D3
- DMACRYS: use of DMACRYS for potential calculations.
- DOINVERSION: forces inversion check of structures for distance
metric in minpermdist, even for biomolecules.
- DONTMOVE n1 n2 … : prevents atoms n1, n2,… moving during MC step taking. They can still move during minimisation.
- DONTMOVEGROUP centre radius type: If type is set as default to GT, DONTMOVEGROUP prevent all atom greater than radius angstroms from the centre atom from moving during MC step taking. type can also be set to LT to not move all atoms within radius of centre.
- DONTMOVERES n1 n2 … : prevents all atoms in residues n1, n2,… from moving during MC step taking.
- DONTMOVEALL n1 n2 … : prevents all atoms from moving during MC step taking (usually used with DOMOVE and DOMOVERES).
- DOMOVE n1 n2 … : allows atoms n1, n2,… to move during MC step taking. Only functions in conjunction with
DONTMOVEALL
- DOMOVERES n1 n2 … : allows all atoms in residues n1, n2,… to move during MC step taking. Only functions in conjunction
with DONTMOVEALL
- DUMP: if present will cause the initial and final energy and quench geometry for every step
to be dumped into dump.X.xyz where X is an integer.
If DEBUG is set then all the structures encountered in the minimisation are also dumped.
The geometries are saved
in XMakemol xyz format. If CHARMM is also specified, dump.pdb and dump.crd
are produced containing each quench geometry in PDB and CHARMM CRD format.
- DUMP_MARKOV nwait nfreq: dump the Markov geometry and
energy (every nfreq trial steps and after the first nwait) to file dump.X.xyz, where X is an integer. Intended for use instead of DUMP and in conjunction with FIXBOTH to facilitate the characterisation of equilibrium structure as part of post-processing.
- DUMPBEST: dump best structures after every step.
- DUMPINT int: changes the default interval for dumping a restart
GMIN.dump file from 1000 basin-hopping steps to int.
- DUMPMIN: When using more than one processor (e.g. BHPT), or in conjunction with DUMPSTRUCTURES, the SAVE lowest minima will be dumped every
DUMPINT steps as dumpmin.x files.
- DUMPQU: when using AMBER9, dumps each quench geometry in rst format to quenchX.rst
and pdb format to quenchX.pdb. Dumping does not occur if a chirality check fails.
- DUMPSTEPS: when using AMBER9, dumps each geometry after the MC step has been taken in rst format to afterstepX.rst
and pdb format to afterstepX.pdb.
- DUMPSTRUCTURES: dump best structures in rst, xyz and pdb format.
- DUMPUNIQUE: dump unique structures as soon as they are found.
- DZUGUTOV dzp1 dzp2 dzp3 dzp4 dzp5 dzp6 dzp7: Dzugutov potential in a general form.
The parameters are m, A, aa, B, d, bb and m2.
- EAMAL: specifies an embedded atom model for aluminium.
- EAMLJ A0 beta Z0: specifies the EAMLJ potential (Baskes, Phys. Rev. Lett.,
27, 2592, 1999) with parameters A0, beta and Z0.
- EDIFF econv: quench minima are only considered to be different if their
energies differ by at least econv. This option mainly affects the lowest energy
saved geometries. If the current quench energy is within econv of a saved energy, but
lies lower, then the saved energy and geometry are replaced.
The default is 0.02 but different values are appropriate for different potentials.
- ENERGY_DECOMP: print energy decomposition for AMBER9 and AMBER12
- ENPERMS: Enumerate all distinct permutations of a binary system (0 <NTYPEA <NATOMS) using Lehmer's algorithm. Instead of a traditional geometry-perturbing step, Lehmer's algorithm will generate a new permutationl isomer by swapping the coordinates of two deterministically-picked unlike atoms. The procedure terminates once all the distinct permutations have been enumerated.
- EQUILIBRATION equil DumpEveryNthQuench: equil is the number of
MC steps preceding the accumulation of the
density of states histogram in a Wang-Landau
basin-sampling run. The default is 0. DumpEveryNthQuench specifies how often the
statistics are recorded into the output files.
- EWALD N ewaldrealc ewaldrecipc : computes long-range potentials (such as electrostatics)
using Ewald summation. N specificies the order of the potential (use 1 for electrostatics).
ewaldrealc and ewaldrecipc are the real-space and reciprocal-space cutoffs. This
should only be used with periodic systems, so the PERIODIC keyword must be specified.
- EXEQ n: for use with the RESERVOIR keyword. After a configuration is
taken from the reservoir by the lowest temperature non-reservoir replica we do not
record visits statistics for n steps. This allows some local equilibration if
the configuration is not truely equilibrium.
- EXPANDRIGID freq factor (NORMALISE): for use with the generalised rigid body framework,
RIGIDINIT. Expands the system by a factor of factor by scaling the distance
of each rigid body from the centre of mass of the system every freq steps. If the third argument NORMALISE
is used, all rigid bodies are simply translated by factor from the system centre of mass.
- EXTRACTFRQS : extract the normal mode frequencies of minimum n from
min.frqs and write them to file extractedfrqs.
For use with USEMINDATA and CNBH.
- EXTRACTMIN n: extract the coordinates of minimum n from
points.min and write them to file extractedmin.
For use with USEMINDATA and CNBH.
- FAL : specifies the Farkas potential for aluminium.
- FASTOVERLAP N σG lmax: Specify the use of the FASTOVERLAP alignment algorithm described in Griffiths et al., JCTC 2017. This algorithm overrides the default MINPERMDIST alignment subroutine. FASTOVERLAP uses the FFTW library to quickly maximise the overlap of Gaussian kernels centred on each atom in the two structures being aligned. It can be used for either periodic or aperiodic systems. Use of FASTOVERLAP is recommended for periodic systems, but for aperiodic it can be very slow, and use of the BRANCHNBOUND keyword is recommended instead. FASTOVERLAP is deterministic and systematically improvable.
The alignment procedure is repeated N times from different starting positions, and the best alignment is used. For periodic systems, starting positions are generated by applying global translational displacements to one of the structures. For aperiodic systems, global rotations are used. N defaults to 1, but larger values can yield improved accuracy for extra computational cost.
σG is the width of the Gaussian kernel used in the alignment. For a discussion of appropriate values, see the original paper. If left unspecified, σG defaults to 1/3 of the average interatomic spacing, which is usually a sensible value. This spacing is determined from the number of particles and box lengths for a periodic system (so the default value will not be suitable if the system is inhomogeneous), and measured directly from the two structures being aligned in the case of an aperiodic system.
lmax is the maximum angular momentum used in SOFT transforms for FASTOVERLAP with aperiodic systems. The default value of 15 should be appropriate for most systems, but a larger value may be required for very large clusters and biomolecules. This parameter has no effect for periodic systems.
FASTOVERLAP is compatible with the OHCELL keyword to test for unit cell symmetries in cubic periodic systems. Note that, by default, FASTOVERLAP will test inversion isomers for alignment, which may not be appropriate for all systems (especially biomolecules). The NOINVERSION keyword should be specified to avoid this.
- FEBH
TFEBH: do a free energy basin-hopping run. This uses the free energy
calculated using the harmonic superposition approximation for the acceptance criterion. This also
leads to rejection of transition states from the Markov chain (GMIN has no in-built check to guarantee
this for a potential energy run). Additionally, it enables the MIN_ZERO_SEP keyword, which
uses the separation of the zero eigenvalues (typically the 6 corresponding to overall translation and
rotation) to determine convergence.
- FIXEDEND : requires documentation.
- FIXBOTH : both the temperature and maximum step size are fixed regardless of
the calculated acceptance ratio.
- FIXCOM : fix centre of mass rather than centre of coordinates.
- FIXCELLANGLES : to be used in combination with BOXDERIV: prevents modification of cell angles during lattice optimisations.
- FIXSTEP : the maximum step size is fixed and the temperature is varied to
try and achieve the requested acceptance ratio.
- FIXTEMP : explicitly fixes the temperature. Only used if FIXSTEP is set, in
which case using FIXTEMP gives a result equivalent to FIXBOTH.
- FNI : specifies the Farkas potential for nickel.
- FORBIDLIST n : steps are forbidden to any structure with an energy within a threshold close enough to any
of the n values specified in file forbid.
The energy tolerance is specified by the EDIFF keyword.
- FRAUSI : specifies a particular tight-binding potential for silicon.
See also keyword ANGSTROM.
- FREEZE n1 n2 … : freeze the coordinates of atoms n1, n2,…. Atoms affected by FREEZE will not move during MC step taking,
or during minimisation as their gradients are set to zero.
Frozen atoms may also be specified in a file frozen. The first line of the file should contain the number of frozen atoms. Subsequent lines contain the atom indices to be frozen.
- FREEZEGROUP centre radius type: If type is set as default to GT, FREEZEGROUP FREEZEs all atom greater than radius angstroms from the centre atom. type can also be set to LT to FREEZE all atoms within radius of centre.
- FREEZERES n1 n2 … : freeze the coordinates of all atoms in residues n1, n2,….
- FREEZEALL n1 n2 … : freeze the coordinates of all atoms
- UNFREEZE n1 n2 … : unfreeze the coordinates of atoms n1, n2,…. Only functions in conjunction with
FREEZEALL
- UNFREEZERES n1 n2 … : unfreeze the coordinates of all atoms in residues n1, n2,…. Only functions in conjunction with
FREEZEALL
- FS gatom: specifies a Finnis-Sinclair potential using parameters from
Finnis and Sinclair, Phil. Mag. A, 50, 45 (1984)
and corresponding erratum Phil. Mag. A, 53, 161 (1986).
gatom=1 for V, gatom=2 for Nb, gatom=3 for Ta, gatom=4
for Cr, gatom=5 for Mo, gatom=6 for W, gatom=7
for Fe (original parameters), gatom=8 for Fe (modified parameters in erratum).
Subtoutine FS was coded by Ja,es Elliott in April 2009.
- GA population offspring generations: specifies optimisation using a
``Lamarckian'' genetic algorithm[#!Johnston03!#] instead of Monte Carlo. If
STEPS is defined, each new member of the population is optimised with a
basin-hopping Monte Carlo search. The search will end when the number
of generations has been exhausted or when solution specified by
the TARGET keyword has been reached. Currently, this is only implemented
for the BLN model protein and clusters. See §3.2 for more
details.
- GABHINCR increment: specifies the use of a hybrid
GA/basin-hopping algorithm with a variable length of basin-hopping search. The
increment defines the number of extra basin-hopping steps to perform in
each generation. This does not have to be an integer (e.g. increment =
0.5 increases the number of basin-hopping steps by one after every other
generation).
- GACROSS n: specifies n-point crossover for genetic algorithm
mating operations. Permitted values for n are 1 and 2 (default = 1).
- GADUMPPOP: dump genomes of entire population after every generation.
- GADUPPRED ethresh: sets the energy threshold for the GA
duplicate predator. If two structures differ by less than ethresh, they are marked as duplicates and the least stable one is
removed from the population.
- GAEPOCH ethresh save DUMP: specifies the use of a new epoch
operator to restart GA searches that have converged. If the mean energy of
the population decreases by less than ethresh in one generation, a new
population of random solutions is generated and a new epoch begins. Optionally,
the best save solutions are carried over from one epoch to the next.
Adding the optional DUMP statement prints the conents of the population to
a file epoch.n at the end of each epoch.
- GAINITCHAIN: random structures at the start of an epoch
in a GA search will be generated as a sequence continuous chain. If
no GAINIT... option is specified, this will be chosen for BLN
proteins.
- GAINITSPHERE radius: random structures at the start of an epoch
in a GA search will be generated as points inside a sphere. If
no GAINIT... keyword is specified, this will be chosen for clusters.
The default radius is 3.0.
- GAMUTRATE mutrate: sets the mutation rate for GA
searches. Default=0.1.
- GASELROUL tournsize: specifies the roulette wheel method to select
parents for mating in GA searches. The probabilities for the roulette
wheel are generated using a tanh fitness function.
- GASELTOURN tournsize: specifies the tournament method to select
parents for mating in GA searches. Default tournsize=3.
- GATWIN: allow twinning moves for GA.
- GBD kappa kappaprime mu nu sigma0 epsilon0: calls a finite system of discoids
interacting via a modified Gay-Berne pair potential due to Bates and Luckhurst [#!BatesL96!#].
- GBH: parallelised generalised basin hopping
- GCBH mu nmax int relax prob: specifies grand canonical basin-hopping.
mu is the chemical potential, nmax is the maximum number of atoms,
int is the interval between changes in the number of atoms,
relax is the block size for relaxation with conventional BH steps,
prob is the probability of increasing the number of atoms by one,
and 1-prob is the probability of removing an atom.
The grand canonical accept/reject is based on the lowest potential obtained within
a Markov chain of blocks, to allow the system to relax when the number of atoms
increases or decreases.
The atom removed is currently programmed to be the one that is most weakly bound, so
this pair energy must be known.
A new atom is added two distance units outside the current cluster defined
by the atom furthest from the centre of coordinates.
In both cases an initial quench is performed immediately, and the attempt is rejected
if this quench is unsuccessful.
Some declarations are based on nmax, while locally dynamic allocations
for subroutines can use the current number of atoms.
If FEBH is specified then the potential is based on the exponential factor
and the partition function (including rotation if USEROT is used).
Otherwise, the test is based on the potential defined by
β(μN - V).
- GEOMDIFFTOL diff: tolerance on distance between two structures to be counted as identical.
- GETCNSIZE tmin tmax tinc mumin mumax muinc maxmin: for use with CNBH.
Find the size corresponding to the estimated critical nucleus over a grid of
temperature and chemical potential values. tmin, tmax, tinc are the minimum, maximum and temperature
increment,
mumin, mumax, muinc are the minimum, maximum and chemical potential increment.
maxmin is the maximum number of minima to use in the superposition partition function
for each size. Minima are taken in the order they are found in min.data.
- GLJ n1 n2 n3 etc.: specifies general Lennard-Jones potential for any number of
differ LJ species.
The ε and σ parameters for all the different species should be specified as
the lower diagonal blocks of the corresponding matrices on the following lines.
For example, to specify a 13-atom cluster with three different species:
GLJ 6 4 3
1.0
1.1 1.2
1.2 1.3 1.4
1.0
1.5 1.6
1.7 1.8 1.9
- GLJY alpha A xi: specifies a combination of generalised Lennard-Jones and Yukawa potentials for colloidal systems.
The potential is given by
U(r) = 4ε
 (σ/r)2α - (σ/r)α)
(σ/r)2α - (σ/r)α)![$\left.\vphantom{ (\sigma/r)^{2\alpha} - (\sigma/r)^{\alpha}) }\right]$](img22.png) + A exp(- r/ξ)/(r/ξ)
in reduced units with
ε = σ = 1.
+ A exp(- r/ξ)/(r/ξ)
in reduced units with
ε = σ = 1.
- GROUND: when combined with keywords NEON or ARGON
uses an accurate (Aziz) potential to model the ground state neutral cluster.
- GROUPROTATION (freq) (scalemode) (offset): specifies group rotation moves for groups of atoms defined in atomgroups. freq
(optional) specified the frequency with which these moves should be made. The default, 1, specified group rotations be made every 1 steps.
scalemode (optional) can be used to determine how the GROUPROTATION moves are scales to attempt to achieve the desired acceptance ratio.
There are 4 options for scalemode - SCALENONE (default): do no scaling, SCALEPROB: scale the group selection probabilities, SCALEROT: scale
the rotation amplitudes and SCALEBOTH: scale both selection probabilities and rotation amplitudes.
offset (optional) can be used for systems with consistant ligand or cofactors to allow the ligand/cofactor group numbering to be
system independant. For example, if there are 3500 atoms in a protein, and the ligand starts at atom 3501, setting offset to 3500 means
that the GROUP in atomgroups numbering starts at 1 again. The default offset is 0. Currently this is only usable with
AMBER9 and CHARMM.
The atomgroups file is formatted as follows:
GROUP name bondatom1 bondatom2 groupsize rotationscalefactor probselect
groupatom1
groupatom2
groupatom3
...
The group rotation axis is defined by the vector from bondatom1->bondatom2, and the rotation is scaled by rotationscalefactor
.
Here is an example atomgroups file containing two groups:
GROUP OME 6 5 4 1.0 0.8
1
2
3
4
GROUP CH2OH 23 25 4 1.0 0.8
26
27
28
29
To create the atomgroups file for proteins with standard amino acids, you can either visualize the molecule with atom labels using any visualization software, such as VMD or PyMOL, and create the file manually, or generate it using the available Python scripts. The relevant scripts can be found in ~/softwarewales/UTILS/GMIN. The file generate_atomgroups.py takes a PDB file as input and generates the corresponding atomgroups file for side-chain torsions. It is compatible with Python 3 and requires the numpy and biopython libraries. If these libraries are not available, you can install these using pip install numpy and pip install biopython, respectively. Note that you do not need to include the .pdb extension when specifying the file in the command. The file works for capped proteins as well. For a file input.pdb, its usage is as follows:
python generate_atomgroups.py input
Let’s consider a simple dipeptide {ARG GLU} as an example:
| ATOM |
1 |
N |
ARG |
1 |
3.326 |
1.548 |
-0.000 |
1.00 |
0.00 |
N |
| ATOM |
2 |
H |
ARG |
1 |
3.909 |
0.724 |
-0.000 |
1.00 |
0.00 |
H |
| ATOM |
3 |
CA |
ARG |
1 |
3.970 |
2.846 |
-0.000 |
1.00 |
0.00 |
C |
| ATOM |
4 |
HA |
ARG |
1 |
3.672 |
3.400 |
-0.890 |
1.00 |
0.00 |
H |
| ATOM |
5 |
CB |
ARG |
1 |
3.577 |
3.654 |
1.232 |
1.00 |
0.00 |
C |
| ATOM |
6 |
HB2 |
ARG |
1 |
2.497 |
3.801 |
1.241 |
1.00 |
0.00 |
H |
| ATOM |
7 |
HB3 |
ARG |
1 |
3.877 |
3.116 |
2.131 |
1.00 |
0.00 |
H |
| ATOM |
8 |
CG |
ARG |
1 |
4.274 |
5.010 |
1.195 |
1.00 |
0.00 |
C |
| ATOM |
9 |
HG2 |
ARG |
1 |
5.354 |
4.863 |
1.186 |
1.00 |
0.00 |
H |
| ATOM |
10 |
HG3 |
ARG |
1 |
3.974 |
5.548 |
0.296 |
1.00 |
0.00 |
H |
| ATOM |
11 |
CD |
ARG |
1 |
3.881 |
5.818 |
2.427 |
1.00 |
0.00 |
C |
| ATOM |
12 |
HD2 |
ARG |
1 |
2.801 |
5.965 |
2.436 |
1.00 |
0.00 |
H |
| ATOM |
13 |
HD3 |
ARG |
1 |
4.182 |
5.280 |
3.326 |
1.00 |
0.00 |
H |
| ATOM |
14 |
NE |
ARG |
1 |
4.540 |
7.143 |
2.424 |
1.00 |
0.00 |
N |
| ATOM |
15 |
HE |
ARG |
1 |
5.152 |
7.375 |
1.655 |
1.00 |
0.00 |
H |
| ATOM |
16 |
CZ |
ARG |
1 |
4.364 |
8.041 |
3.389 |
1.00 |
0.00 |
C |
| ATOM |
17 |
NH1 |
ARG |
1 |
3.575 |
7.808 |
4.434 |
1.00 |
0.00 |
N |
| ATOM |
18 |
HH11 |
ARG |
1 |
3.089 |
6.925 |
4.509 |
1.00 |
0.00 |
H |
| ATOM |
19 |
HH12 |
ARG |
1 |
3.465 |
8.514 |
5.148 |
1.00 |
0.00 |
H |
| ATOM |
20 |
NH2 |
ARG |
1 |
5.006 |
9.201 |
3.287 |
1.00 |
0.00 |
N |
| ATOM |
21 |
HH21 |
ARG |
1 |
5.605 |
9.375 |
2.492 |
1.00 |
0.00 |
H |
| ATOM |
22 |
HH22 |
ARG |
1 |
4.892 |
9.903 |
4.004 |
1.00 |
0.00 |
H |
| ATOM |
23 |
C |
ARG |
1 |
5.486 |
2.705 |
-0.000 |
1.00 |
0.00 |
C |
| ATOM |
24 |
O |
ARG |
1 |
6.009 |
1.593 |
-0.000 |
1.00 |
0.00 |
O |
| ATOM |
25 |
N |
GLU |
2 |
6.191 |
3.839 |
-0.000 |
1.00 |
0.00 |
N |
| ATOM |
26 |
H |
GLU |
2 |
5.715 |
4.730 |
-0.000 |
1.00 |
0.00 |
H |
| ATOM |
27 |
CA |
GLU |
2 |
7.640 |
3.839 |
-0.000 |
1.00 |
0.00 |
C |
| ATOM |
28 |
HA |
GLU |
2 |
8.004 |
3.325 |
0.890 |
1.00 |
0.00 |
H |
| ATOM |
29 |
CB |
GLU |
2 |
8.189 |
3.127 |
-1.232 |
1.00 |
0.00 |
C |
| ATOM |
30 |
HB2 |
GLU |
2 |
7.841 |
2.094 |
-1.241 |
1.00 |
0.00 |
H |
| ATOM |
31 |
HB3 |
GLU |
2 |
7.841 |
3.636 |
-2.131 |
1.00 |
0.00 |
H |
| ATOM |
32 |
CG |
GLU |
2 |
9.698 |
3.149 |
-1.195 |
1.00 |
0.00 |
C |
| ATOM |
33 |
HG2 |
GLU |
2 |
10.047 |
4.181 |
-1.186 |
1.00 |
0.00 |
H |
| ATOM |
34 |
HG3 |
GLU |
2 |
10.047 |
2.640 |
-0.296 |
1.00 |
0.00 |
H |
| ATOM |
35 |
CD |
GLU |
2 |
10.248 |
2.436 |
-2.429 |
1.00 |
0.00 |
C |
| ATOM |
36 |
OE1 |
GLU |
2 |
10.455 |
3.138 |
-3.454 |
1.00 |
0.00 |
O |
| ATOM |
37 |
OE2 |
GLU |
2 |
10.455 |
1.197 |
-2.334 |
1.00 |
0.00 |
O |
| ATOM |
38 |
C |
GLU |
2 |
8.188 |
5.259 |
-0.000 |
1.00 |
0.00 |
C |
| ATOM |
39 |
O |
GLU |
2 |
7.425 |
6.222 |
0.000 |
1.00 |
0.00 |
O |
| TER |
|
|
|
|
|
|
|
|
|
|
| END |
|
|
|
|
|
|
|
|
|
|
The associated atomgroups file for side-chain torsions thus obtained is given as:
GROUP ARG1CACB 3 5 17 0.25 0.25
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
GROUP ARG1CBCG 5 8 14 0.25 0.25
9
10
11
12
13
14
15
16
17
18
19
20
21
22
GROUP GLU2CACB 27 29 8 0.25 0.25
30
31
32
33
34
35
36
37
GROUP GLU2CBCG 29 32 5 0.25 0.25
33
34
35
36
37
Similarly, to generate an atomgroups file corresponding to backbone dihedrals, a similar procedure can be followed using ~/softwarewales/UTILS/GMIN/generate_dihedral_atomgroups.py. For the above mentioned file input.pdb, its usage is as follows:
python generate_dihedral_atomgroups.py input, which generates:
GROUP ARG1psi 3 23 16 0.25 0.1
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
GROUP GLU2phi 25 27 12 0.25 0.1
28
29
30
31
32
33
34
35
36
37
38
39
- QUIETGROUPROT: suppresses output from group rotation about which angles were changed.
- GTHOMSON type param1 param2 param3 param4: the system will be constrained to a curved surface.
The type of surface is specified by type as follows:
1 A cylinder, with height param1 and radius param2.
2 A catenoid
3 An unduloid
4 An unduloid
5 A sphere, with radius param1.
6 A Möbius strip, with radius param1 and width param2.
- GTHOMSONPOT type σ ρ: specifies the potential to use with the surface described by
the GTHOMSON keyword. The values of type give the following behaviours:
1 A Coulomb potential, with all particles having a charge of 1.
2 A
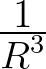 potential.
potential.
3 A Yukawa potential, with screening length σ.
4 A Lennard-Jones potential, with characteristic distance σ and a well depth of 1.
5 A repulsive Lennard-Jones (
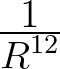 ) potential,
with characteristic distance σ.
) potential,
with characteristic distance σ.
6 A Morse potential, with characteristic distance σ and range parameter ρ.
- GUIDE guidecut: specifies the RMS force below which the real potential is used
rather than a guiding potential. The systems affected are CPMD and WELCH,
which are guided by AMBER and TOSI, respectively, and also PACHECO,
where the Axilrod-Teller contribution is only included when the RMS force falls below
guidecut. Default guidecut=0.0001.
New guided potentials are ZETT1 and ZETT2 (guided by Morse with ρ = 5) and
NATB (guided by GUPTAT). Parameters for the guiding potential must also be specified in
data.
- GUIDECASTEP system runparams: specifies that xtb should be used as the guiding potential for CASTEP.
The strings following GUIDECASTEP are the same as for the XTB keyword. A file system
is needed in the current working directory. A coords file is not required because the
coordinates are read from a CASTEP cell file. Example lines from data:
GUIDECASTEP pentaprismane '–acc 0.1 –gfn1 -P 10 –norestart'
GUIDE 0.001
- GUIDESYM rho eps conv group: specifies a guiding potential that aims to symmetrise the
system. rho > 0 is a range parameter, eps > 0 multiplies the whole symmetrising function, which is subtraced
from the principal potential, conv is the RMS convergence condition for switching off the guiding term,
and group is the point group to use.
This keyword uses an exponential in the continuous symmetry measure.
If conv < 0 the guiding potential is always on, and this parameter is otherwise ignored.
- GUIDESYM2 rho eps conv group: is the same as GUIDESYM but the continuous
symmetry measure is based on an inverse squared distance instead of using an exponential.
- GUPTA gatom: specifies a Gupta potential using parameters from Cleri and Rosato,
Phys. Rev. B, 48, 22 (1993). gatom=1 for Ni,
gatom=2 for Cu,
gatom=3 for Rh,
gatom=4 for Pd,
gatom=5 for Ag,
gatom=6 for Ir,
gatom=7 for Pt,
gatom=8 for Au,
gatom=9 for Al,
gatom=10 for Pb,
gatom=11 for Ti type 1,
gatom=12 for Ti type 2,
gatom=13 for Zr type 1,
gatom=14 for Zr type 2,
gatom=15 for Co,
gatom=16 for Cd type 1,
gatom=17 for Cd type 2,
gatom=18 for Zn,
gatom=19 for Mg,
gatom=20 for V,
gatom=21 for Na,
gatom=22 for Sr (Wang and Blaisten-Barojas, J. Chem. Phys., 115, 3640 (2001)),
gatom=22 for Au as used by Garzon et al.
The Gupta subroutine was recoded more efficiently by James Elliott in April 2009.
- HBONDLIGAND (ligandresn): used with the HBONDMATRIX keyword. When specified, groups are defined only
by the hydrogen-bonds between the ligand and protein only - not between sidechains. The optional arguement ligandresn
specifies which line in the residuefile corresponds to the ligand. The default is to assume it is the last line of the file.
This keyword must be specified after HBONDMATRIX in the data file.
- HBONDMATRIX donoracceptorfile residuefile mode: used with the AMBER keyword to focus GMIN sampling on
the diversity of ligand binding-modes in a protein/ligand system. donoracceptorfile should contain the AMBER ptraj
definitions of the hydrogen-bond donor and acceptor atoms for your system as described in the AMBER Tools manual (see Google).
residuefile should contain the list of residue numbers (one per line) that are involved in binding. When you have no
specific information, this can just mean those in close proximity to the binding site. mode is an optional arguement, which
defaults to ACCEPT. If REJECT is used instead, HBONDMATRIX will instead restrict sampling to the binding mode identified after
the initial quench. This can be used to optimise a single binding mode.
- HBONDSOFTCUT dloose dtight aloose atight: used with HBONDMATRIX to specify a custom cutoff window for the
hydrogen-bond analysis used by the HBONDMATRIX keyword to group structures. The default values for the four parameters are
3.05, 2.95, 115.0 and 120.0. It should be noted that the actual angles used in the analysis are π -aloose and
π -atight. This is why the looser angle cutoff is actually numerically smaller.
- HYBRIDMIN rigidconv: enables hybrid rigid body/all-atom minimisation when using RIGIDINIT. rigidconv is
the RMS force convergence criterion for the rigid body minimisation. Once converged, an all-atom minimisation begins using the
convergence specified in SLOPPYCONV. The final quenches are done atomistically.
- INTMIN: used with CHARMM keyword to specify minimisation in internal
coordinates. This generally appears to be
slower than using Cartesian coordinates.
- INVERTP: specifies the inverted potential. The energy and all derivatives
are multiplied by -1.
This converts searches for the highest index saddles into minimisation.
However, it is only likely to work for bounded potentials.
- IONICCOLLOID pos prad nrad ppot npot: specifies the ionic colloid potential from
Nature, 580, 487, 2020.
Particle positions should be in nm with the pos positively charged particles first.
prad and nrad are the radii of the positive and negative particles in nm,
and their surface potentials in millivolts are ppot and npot.
The following parameters are set in the corresponding subroutine:
brush density 0.09 nm-2,
brush length 10 nm,
debye length 5.25 nm
dielectric constant 80,
temperature 298 K.
- JC: Specifies Murrell's two- plus three-body
potential.[#!murrellm90!#,#!murrellr90!#,#!alderzijmr91!#,#!eggenjlm92!#,#!fengjm93!#]
A file JMparams must
exist in the current directory containing the parameters
c0, c1,…, c10, re,I>D, a2 and a3. An optional cutoff parameter can also be provided at the end of the
JMparams file.
Subroutines used: jmec, jm2c, jm3c.
- JUMPMOVE np1 np2 int: specify J-walking type attempts between parallel runs np2
and np1 at intervals of int steps.
- LAMMPS datafile: invokes the LAMMPS interface, where energies and gradients are obtained from
the LAMMPS package. This keyword requires file in.lammps in the working directory to
set up the LAMMPS input and the initial coordinates.
If the option parameter datafile is provided then GMIN will know the lammps data file referred to
in in.lammps without needing to try and parse using grep and sed.
It also requires the MPI keyword, since the LAMMPS interface assumes an MPI compilation for LAMMPS.
The current build has been tested on sinister and nest for branch DJWlammps, and has hardcoded libraries.
It works with modules cmake/3.16.3, gcc/7.5.0, mpi/openmpi/gnu7/4.1.0, and the cmake line is
FC=mpif90 CC=mpicc CXX=mpicxx cmake -DWITH_LAMMPS=1 ~/softwarewales/GMIN/source/ -DCOMPILER_SWITCH=gfortran
The lammps_mpi branch works with modules
gcc/7.5.0 mpi/openmpi/gnu7/4.1.0 cmake/3.16.3
and
FC=mpif90 CC=mpicc CXX=mpicxx cmake /softwarewales/GMIN/source -DCOMPILER_SWITCH=gfortran -DWITH_LAMMPS=yes -DWITH_ALIGN=no -DWITH_MYBLAS=yes -DWITH_MYLAPACK=yes
The lammps_update branch provides an alternative more recent interface based on OpenMP, where standard compilation should work.
See LAMMPSMDSTEP below for alternative basin-hopping step perturbations.
- LAMMPSMDSTEP mdsteps mdtemp:
this keyword is currently available only on the lammps_update branch.
It allows basin-hopping geometry perturbation steps based on NVT or NPT molecular dynamics runs supplied by lammps,
with mdsteps in each molecular dynamics run.
The current implementation uses new random velocities created for each run at temperature
mdtemp, to prevent identical steps from the same minimum.
Be warned that lammps is likely to stop if the step size or temperature is too large. GMIN will attempt to
reinitialise lammps if such errors are detected.
The other molecular dynamics parameters, including temperature and thermostat and barostat damping
coefficients, are read from the in.lammps file. For the NPT ensemble the box lengths will
change (by default isotropically), but it is not possible to set them directly without reinitialising lammps.
GMIN therefore performs constant volume geometry optimisation at the current box parameters, and saves the box lengths
with the energy and configuration. The final quench phase is skipped. To help with restarting an npt run
GMIN will attempt to parse the in.lammps file to discover the name of the lammps data file, and
if successful will produce a restart.data.n and a coords.n file for the saved minima, with
the appropriate box lengths in restart.data.n. If the data file name is provided on the
LAMMPS keyword line in the data file GMIN will not need to look for it in
in.lammps.
The coordinates in coords.n are the same as in
the lowest and are just provided for convenience. The initial quench in such a run should agree
with the minimum that was obtained in the parent run.
GMIN looks for a read_data line in the in.lammps file and uses various grep and sed system commands
to get the file name. For this approach to work the in.lammps file needs to avoid tab characters
and commented additional read_data lines.
The lammps_update branch provides an alternative more recent interface based on OpenMP, where standard compilation should work.
See LAMMPSMDSTEP below for alternative basin-hopping step perturbations.
- LAMMPSMDSTEP mdsteps mdtemp:
this keyword is currently available only on the lammps_update branch.
It allows basin-hopping geometry perturbation steps based on NVT or NPT molecular dynamics runs supplied by lammps,
with mdsteps in each molecular dynamics run.
The current implementation uses new random velocities created for each run at temperature
mdtemp, to prevent identical steps from the same minimum.
Be warned that lammps is likely to stop if the step size or temperature is too large. GMIN will attempt to
reinitialise lammps if such errors are detected.
The other molecular dynamics parameters, including temperature and thermostat and barostat damping
coefficients, are read from the in.lammps file. For the NPT ensemble the box lengths will
change (by default isotropically), but it is not possible to set them directly without reinitialising lammps.
GMIN therefore performs constant volume geometry optimisation at the current box parameters, and saves the box lengths
with the energy and configuration. The final quench phase is skipped. To help with restarting an npt run
GMIN will attempt to parse the in.lammps file to discover the name of the lammps data file, and
if successful will produce a restart.data.n and a coords.n file for the saved minima, with
the appropriate box lengths in restart.data.n.
If the data file name is provided on the
LAMMPS keyword line in the data file GMIN will not need to look for it in
in.lammps.
The coordinates in coords.n are the same as in
the lowest and are just provided for convenience. The initial quench in such a run should agree
with the minimum that was obtained in the parent run.
GMIN looks for a read_data line in the in.lammps file and uses various grep and sed system commands
to get the file name. For this approach to work the in.lammps file needs to avoid tab characters
and commented additional read_data lines.
- LB2: specifies the potential[#!LB299a!#,#!LB299b!#,#!LB204!#]
where ε and σ are set to unity.
- LFLIPS n m kT (mfac): for every nth step in the main basin-hopping sequence perform a subsequence of m steps with flip moves only. This subsequence constitutes an independent block of semi-grand canonical basin-hopping at a given kT, with the total number of atoms fixed but the relative population of constituent species allowed to vary. In every instance the subsequence starts with the specified value of kT, which is then multiplied by the (optional) parameter mfac on each of the m successive steps. (Default: mfac = 1.) The keyword SEMIGRAND_MU can be used to impose non-zero semi-grand chemical potentials (with respect to the first species).
- LFLIPS_RESET: at the start of every subsequence of flips, the stoichiometry will be reset to the value inferred from the keyword specifying the multicomponent potential. The atomic labels will be reassigned randomly.
- LIGMOVE ligrotscale ligcartstep ligtransstep ligmovefreq: used with AMBER9 and MOVABLEATOMS. Specifies ligand only rotation, cartesian perturbation and translation. The ligand is defined by atom index in the file 'movableatoms'. Setting ligrotscale less than 1.0
limits the ammount of rotation possible - this may be required to prevent cold fusion with non-spherical ligands. ligcartstep and ligtransstep
define the maximum size (in angstroms) of the random cartesian perturbations and rigid body translation applied to the ligand respectively.
ligmovefreq can be set to greater than 1 to prevent ligand moves being applied every step i.e. ligmovefreq = 2 for every other step.
All ligand moves are applied AFTER any MD if AMBERMDMOVES is on to prevent the MD exploding.
- LJADD: specifies an addressable LJ potential. A data file epsilon is required
specifying the well depth parameter ε for all pairs of atoms, one per line.
Choosing values according to nearest neighbours in a reference geometry can bias the
system towards that configuration.
- LJADD2 n: specifies an addressable LJ potential for multiple copies of an n-particle
cluster. The data file epsilon is required
with n×n entries, one per line. These well depth parameters are then applied modulo n.
For particles with the same address, only the repulsive part of the LJ potential is used.
- LJAT Z* scale: specifies the Lennard-Jones plus Axilrod-Teller
potential for a reduced triple-dipole strength of Z*.
Provided mostly as a guiding function for DFTBC.
rescale is the coordinate rescaling factor from the LJAT potential
to DFTB, default value 2.424.
- LJCOUL nc q' f
Tswap specifies a cluster of Lennard-Jones particles in which the first nc
particles carry identical reduced charges q' in addition to the Lennard-Jones interaction.
The parameter f specifies what fraction of the Monte Carlo steps should be swaps between the
positions of a charged and a neutral particle, rather than a conventional step.
Tswap is the temperature to be used in the acceptance criterion for swap moves, overriding
that specified using the TEMPERATURE keyword. Generally, a lower temperature is more effective
at finding the lowest-energy permutation of charges. The default value of
Tswap is zero.
The reduced charge q' is related to the actual charge q by
q' = q/(4πε0εσ)1/2,
where ε and σ are the Lennard-Jones well depth and length parameter respectively.
This way, the reduced energy of two charges is
E' = q'2/r', where
r' = r/σ is the reduced distance
bdetween the charges.
- LJCOULADD3: a modification of the LJ potential coded into CUDA, which Coulombic and harmonic restraint
functions are added. It only works with CUDA and needs to be used in conjunction with SANDBOX. The necessary files are
rbdata and coords. It populates the arrays of interparticle interaction strengths from the interaction matrix in the
rbdata file. If use LJADD3 and SANDBOX keywords, they should be defined before the LJCOULADD3 keyword.
- LJGAUSS mode rcut ε r0 σ2 params specifies that the Lennard-Jones Gauss double well potential should be used.
The Lennard-Jones potential has an energy minimum of 1 and a minimum at distance 1.
The relative weight of the Gaussian potential is ε and the Gaussian peak is at distance r0. The width of the Gaussian is
controlled by σ2. The Stoddard-Ford procedure is used to make the potential and its first derivatives go smoothly to zero at rcut.
mode can be either 0, 1, 2 or 3. mode 0 specifies the normal potential, and can be either a cluster or with periodic
boundary conditions with an orthogonal unit cell using the PERIODIC keyword. mode 1 or 2 requires periodic boundary conditions.
The PERIODIC keyword must be used, but the unit cell parameters from it will be ignored. Instead, the last line of the coords file will
be interpreted as the unit cell length parameters and will be optimised. Additionally, for mode 2, the second
last line of the coords file be interpreted as unit cell angle parameters for a triclinic unit cell, and optimised. Since there
are combinations of unit cell angles that are physically impossible, steps will be rejected if such combinations are detected.
In mode 3, as well as the cell being optimised, potential parameters specified by params (an integer between 1 and 7)
will also be optimised. If the 1 bit of params is set, ε will be optimised, if the 2 bit is set, r0 will be optimised,
and if the 4 bit is set, σ2 will be optimised.
- LJPOLY length sig eps k bond: Lennard-Jones chain polymers each with length atoms,
harmonic springs between successive atoms and LJ terms between
non-bonded atoms. sig and eps are the usual LJ σ and ε parameters, bond is the harmonic spring
equilibrium distance, and k is half the spring force constant. The half is for compatibility with the LAMMPS harmonic bond setup.
See also LJRING.
- LJPOLYSTEP: take the atomic Cartesian displacements along the spring direction to the next
bonded atom (or the repvious one for the end of a chain). This step is for use with LJRING and LJPOLY
to try and prevent topology changes. The maximum value for STEP should not be more than the equilibrium bond length.
It may be better to combine this step with a compression move using COMPRESS and GUIDE.
- LJSPRINGS sig eps k bond: general Lennard-Jones plus springs. Requires a file springfile
with the bonded pairs, one pair per line. The Lennard-Jones terms are applied between all non-bonded pairs with
σ and ε values sig and eps. The harmonic springs have spring constant 2k and
equilibrium distance bond.
- LJRING length sig eps k bond: Lennard-Jones chain ring polymers each with length atoms,
harmonic springs between successive atoms in a closed chain, and LJ terms between
non-bonded atoms. sig and eps are the usual LJ σ and ε parameters, bond is the harmonic spring
equilibrium distance, and k is half the spring force constant. The half is for compatibility with the LAMMPS harmonic bond setup.
See also LJPOLY.
- LOCALSAMPLE abthresh acthresh: Keyword currently under construction! For three groups of atoms defined in movableatoms
(A,B,C), a step is quenched when the centre of coordinates of A->B is less than abthresh AND A->C is less than acthresh.
If this condition is broken AFTER the quench, it is automatically rejected.
- LPERMDIST n thresh cut orbittol: provides the preferred local
permutational alignment procedure.
n is the maximum number of extra atoms to include in running
minperm for each permutable group (default 11, a value that may be needed to treat phenylalanine), rather than
treating the whole system at the same time, as for PERMDIST.
The extra atoms that define the neighbourhood of the permutable group are
added one at a time in order of their distance from the centre of coordinates of the
group, averaged over the start and finish geometries.
If the optimal aligned distance is greater than thresh (default value 0.2)
then the atom is not included in the neighbour list.
This procedure continues until there are n atoms in the neighbour list,
or no more atoms within the cutoff distance cut, default 10.0.
The parameter orbittol is the distance tolerance for assigning atoms to orbits
in the myorient standard orientation routine, default value 0.001.
This keyword requires the auxiliary file perm.allow to specify permutable atoms, otherwise
all atoms are assumed to be permutable. The absence of a perm.allow
file is considered a mistake for CHARMM runs.
- LSWAPS n m kT (mfac): for every nth step in the main basin-hopping sequence perform a subsequence of m steps with exchange moves only. The keyword is intended for homotop optimisation at regular intervals for a given kT. In every instance the subsequence starts with the specified value of kT, and this value is multiplied by the (optional) parameter mfac on each of the m successive steps. (Default: mfac = 1.)
- MAISE: Use the MAISE library (https://github.com/maise-guide/maise) as the potential. A file callled model must be present in the working directory, that contains the description of the neural net in the format MAISE expects.
- MAKEOLIGO START
nfixnmoveafirst1alast1phimin1phimax1 dmin dmax SCONLY
specifies the oligomer generation procedure. Here, the sample input follows for the generation of a dimer.
The argument START specifies that a new oligomer is to be generated.
The second and third arguments determine how many peptide chains are fixed (nfix) and relocatable (nmove), respectively.
The input geometry has
to be provided in such a form that all fixed peptide chains come first, followed by the peptide chains,
which are set to
new positions during the oligomer generation procedure. The secondary structures of the relocatable peptides are determined via the input
geometry. The following 4⋅nmove arguments specify the first and last atom for each of the relocatable peptide chains, and the
minimum and maximum angle between which the relocatable peptide in question is to be positioned in the xy-plane:
afirsti, alasti, phimini, phimaxi with
i = 1,…, nmove.
The next two arguments determine the minimum and maximum distances, dmin and dmax, which define the boundaries
for the relocation of the peptide chains with respect to the centre of mass of the fixed part of the input structure.
The effect of the last argument SCONLY is that during the subsequent optimisation of the oligomer only the dihedral angles
of the sidechains are perturbed. If this argument is not present, the backbone dihedrals are also changed.
The MAKEOLIGO keyword also affects the rigid body translation and rotation during the subsequent optimisation. The translation is
only performed in the xy-plane. The rotation can be performed either around the z-axis only or in the in the full three-dimensional
space, depending on whether the first argument is START (as in this example) or INITROT. The arguments following
INITROT are identical to those following START.
- MAXBFGS max: max is the largest permitted LBFGS step.
- MAXERISE maxez: specifies the largest rise in energy permitted during an LBFGS
minimisation. MAXERISE must to be large enough for discontinuities encountered
during quenches to be ignored, or the quench may fail. The default is
10-10, or 10-3, for periodic boundary conditions, where
discontinuities can arrise due to multiple images.
- MAXIT maxit maxit2: maxit and maxit2 are integers specifying the
maximum number of iterations allowed in the conjugate gradient quenches. maxit applies
to the `sloppy' quenches of the basin-hopping run and maxit2 to the final quenches
that are used to produce the output in file lowest.
- MD nsteps (nwait nfreq): Perform nsteps molecular dynamics (MD) time steps instead of basin-hopping. Current implementation works only for Cartesian coordinates (not the rigid-body framework), and the atomic positions are dumped to dump.1.xyz every nfreq steps after the first nwait steps, with nwait = nsteps by default (i.e. no dumping). Time-stepping for each coordinate is carried out using the velocity Verlet scheme with a Langevin thermostat:
 |
= |
(1 - γ ) ) + + 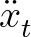 + + 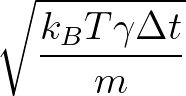 ζ1 ζ1 |
|
| xt+Δt |
= |
xt + Δt |
|
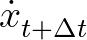 |
= |
(1 - γ) + 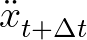 + ζ2 + ζ2 |
|
where x, 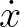 and
and 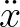 are the coordinate, velocity and acceleration, respectively, with the subscript specifying the time instance; Δt is the time step (see MDPARAMS); m is the atomic mass (see SPECMASS); γ is a damping parameter (see MDPARAMS); and ζ1 and ζ2 are (independent) random numbers from the normal distribution with mean 0 and variance 1. The instantaneous acceleration/force is derived from the potential; the initial coordinates are read from the coords file; the velocities are initialised randomly using the Maxwell distribution, with kBT set by the TEMPERATURE keyword and net translation and rotation subtracted.
are the coordinate, velocity and acceleration, respectively, with the subscript specifying the time instance; Δt is the time step (see MDPARAMS); m is the atomic mass (see SPECMASS); γ is a damping parameter (see MDPARAMS); and ζ1 and ζ2 are (independent) random numbers from the normal distribution with mean 0 and variance 1. The instantaneous acceleration/force is derived from the potential; the initial coordinates are read from the coords file; the velocities are initialised randomly using the Maxwell distribution, with kBT set by the TEMPERATURE keyword and net translation and rotation subtracted.
- MDPARAMS tstep gamma: Specifies the timestep (tstep =
0.002 by default, with units determined by the potential) and Langevin damping
parameter (gamma = 0.0 by default, with units of inverse time). The
default values are tailored for microcanonical MD of atoms with unit mass. For
canonical MD one should choose gamma > 0 such that
0 < gamma*tstep < 1, e.g. gamma of around 10 is
adequate for the default time step.
- MERGEDB dir: merge the min.data, points.min, min.frqs
files from directory dir with
those in the current directory. Common stationary points are identified and
not repeated. Merged results are created corresponding to all the above files.
- MGGLUE: specifies a glue potential for magnesium
- MGUPTA nspecs A_11 p_11 q_11 xi_11 r0_11: specifies a multicomponent Gupta potential for nspecs distinct metallic species. If nspecs = 1 then this single line is sufficient. The model parameters A_11, p_11, q_11, xi_11 and r0_11 have the same meaning as for the keyword BGUPTAT. If nspecs > 1, then
nspecs×(nspecs + 1)/2 - 1 more subsequent lines of the form MGUPTA (n_I) A_IJ p_IJ q_IJ xi_IJ r0_IJ must be supplied for all the remaining I,J ∈ [1,nspecs] in ascending order with J≥I. Note that n_I is expected only when J = I, with n_I specifying the number of atoms for each species I > 1. The value for n_1 (= n_A) is inferred from the knowledge of the total number of atoms (NATOMS). For example, a ternary system AN-k-lBkCl (N = NATOMS) should be specified in six consecutive lines:
| MGUPTA |
3 |
A_AA |
p_AA |
q_AA |
xi_AA |
r0_AA |
| MGUPTA |
|
A_AB |
p_AB |
q_AB |
xi_AB |
r0_AB |
| MGUPTA |
|
A_AC |
p_AC |
q_AC |
xi_AC |
r0_AC |
| MGUPTA |
k |
A_BB |
p_BB |
q_BB |
xi_BB |
r0_BB |
| MGUPTA |
|
A_BC |
p_BC |
q_BC |
xi_BC |
r0_BC |
| MGUPTA |
k |
A_CC |
p_CC |
q_CC |
xi_CC |
r0_CC |
- MIE_FIELD filename Rc Bx By Bz: specifies a fixed substrate from superposition of Mie-type central force fields, each defined by
The parameters n, m, ε, σ and coordinates of each Mie site must be provided in the file filename. The expected file format is:
Nsites n m ε1 ... εNspecies σ1 ... σNspecies
| x1 |
y1 |
z1 |
 |
|
|
|
xNsites |
yNsites |
zNsites |
where ε and σ are given for each species (e.g. for nanoalloys). Mie field(s) can be smoothly truncated using the optional cut-off parameter Rc, which will trigger the usual Stoddard and Ford procedure:
φ(
r)
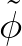
(
r) =
φ(
r) -
φ(
Rc) - (
r -
Rc)
φ'(
Rc).
Periodic boundary conditions with the nearest image convention can be imposed on the field sites using the optional parameters Bx, By and Bz, which specify the periodic box dimensions.
- MINIMUMIMAGE: specifies that the atomic coordinates should be set to the minimum image cell
for periodic calculations.
- MIN_ZERO_SEP separation attempts: when used in combination with the FEBH keyword, this will tighten the rms force convergence thresholds until
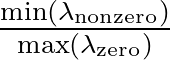 > k where k is the value given by separation. If attempts is specified, the GMIN run will
terminate if the separation is not achieved within the specified number of attempts. This is usually indicative of a problem with the potential or eigenvalue calculation.
> k where k is the value given by separation. If attempts is specified, the GMIN run will
terminate if the separation is not achieved within the specified number of attempts. This is usually indicative of a problem with the potential or eigenvalue calculation.
- MLJ nspecs eps_11 sig_11: specifies a multicomponent Lennard-Jones potential for nspecs distinct species. If nspecs = 1 then this single line is sufficient, and it ought to yield the same results as the standard Lennard-Jones. Extension to nspecs > 1 is analogous to the MGUPTA keyword, and it differs from the input format expected by the GLJ keyword. Note that, although GLJ and MLJ link to different implementations of the general multicomponent Lennard-Jones, they ought to yield the same results.
- MLMIN in start hidden out data λ min: calculates probability and classification properties for min neural network minima
found already. Each neural network is an MLPVB3 network (see below), and the parameters have the same meaning. The weights of each neural network
should be located in a file called MLMINweights, which has the format:
| <weight1,minimum1> |
| <weight2,minimum1> |
| <weight3,minimum1> |
| ... |
| <weight1,minimum2> |
| <weight2,minimum2> |
| <weight3,minimum2> |
| ... |
- MLOWEST: Like TARGET. Accepts multiple target energies and will stop upon hitting *all* targets as opposed to *any* target.
- MLPNORM: Used in combination with any of the machine learning keywords. It specifies that the input features should each be rescaled by the
mean of each feature. This process can help with convergence issues in machine learning. This keyword must be placed in the data file before
the corresponding machine learning keyword, otherwise an error will be thrown.
- MLPVB3 in start hidden out data λ: specifies a perceptron neural network with
one input layer of in input nodes, one hidden layer of hidden hidden nodes,
and one output layer of out output nodes. data is the number of data points
the model trains with, stored in a file called MLPData. This file should be
formatted as follows:
| y1 |
x11 |
x21 |
x31 |
... |
| y2 |
x12 |
x22 |
x32 |
... |
| y3 |
x13 |
x23 |
x33 |
... |
| ... |
|
|
|
|
|
yNdata |
x1Ndata |
x2Ndata |
x3Ndata |
... |
where yi is the outcome for data point i, and xji is the jth input
feature for data point i. The features will be read starting from the
feature in column start (ignoring the y column).
The hidden and output layers also have bias weights contributing to them.
The activation function for the hidden layer is hyperbolic tan.
The activation function for the output layer is just the sum.
Overall, the raw contribution from data point d for output i is given by
yid(;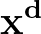 ) = wibo + ) = wibo + 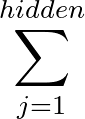 wij(1)tanh wij(1)tanh wjbh + wjbh +  wjk(2)xkd wjk(2)xkd |
(5) |
These outputs become normalised softmax probabilities:
The loss function to be minimised is:
E(;) = - 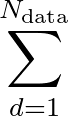 ln pc(d)d(;) + λ ln pc(d)d(;) + λ , , |
(7) |
where c(d ) is the class label for data point d specified by the training set
and λ is a constant coefficient of L2 regularisation, used to prevent overfitting.
All weights are included in this L2 regularisation term, unless the keyword NOREGBIAS is present,
in which case the bias weights are excluded from regularisation.
The number of variables is hidden×(in+out+1)+out.
The variables are ordered
|
wjk(2) |
input to hidden weights (iterating over inputs first), |
|
wij(1) |
hidden to output weights (iterating over hidden first), |
|
wjbh |
hidden bias weights, |
|
wibo |
output bias weights. |
There are other, older keywords MLP3 and MLPB3,
which were similar but had fewer weights. MLP3 had no bias weights.
MLPB3 (now retired) was the same as MLP3 but with a single extra
bias weight added before the tanh activation function in the hidden layer.
Note that MLP3 and MLPB3 did not have the start
parameter available to specify, and the MLPdata file had a slightly different
format: features listed before outcomes.
- MLSUP in start hidden out data λ: specifies a perceptron neural network similar to the MLPVB3 keyword above.
The parameters all have the same meaning as above.
This keyword must come after the MLMIN keyword in the data file.
The number of inputs, start position, and number of data points must be the same as those used for MLMIN.
The number of hidden nodes and the value of λ may be different.
The number of output nodes must be the same as the number of neural network minima given to MLMIN.
Each output is the softmax probability that the neural network minimum corresponding to that output is the best one to use for a given data point.
In this way, MLSUP creates a ``neural network of neural networks":
overall there is a network of weights,
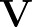 used in a single-hidden-layered neural network, which are used to determine which
neural network with weights should be used for a given data point.
used in a single-hidden-layered neural network, which are used to determine which
neural network with weights should be used for a given data point.
- MORSE rho: specifies a Morse potential
with range parameter rho.[#!braierbw90!#,#!doyewb95!#,#!doyew96a!#]
- MPI: specifies an MPI parallel job.
(only available if the source is compiled with MPI enabled).
- MSC nspecs nn11 mm11 sig11 sceps11 scc1: specifies a multicomponent Sutton-Chen potential for nspecs distinct metallic species. If nspecs = 1 then this single line is sufficient, and it can be used in conjunction with keywords PERIODIC and CUTOFF. The parameters nn11, mm11, sig11, sceps11 and scc11 have the same meaning as for the keyword SC. If a cutoff has been specified, then the potential will be smoothly truncated by a generalised Stoddard and Ford procedure, which ensures the energy and its first derivatives remain continuous. If nspecs > 1, then
nspecs×(nspecs + 1)/2 - 1 more subsequent lines of the form MSC (ntypeI) nnIJ mmIJ sigIJ scepsIJ (sccI) must be supplied for all the remaining I,J ∈ [1,nspecs] in ascending order with J≥I. The parameters ntypeI and sccI are expected only when J = I, with ntypeI specifying the number of atoms for each species I ≠ 1. The value for ntype1 (= ntypeA) is inferred from the knowledge of the total number of atoms (NATOMS). For example, a ternary system AN-k-lBkCl (N = NATOMS) should be specified in six consecutive lines:
| MSC |
3 |
nnAA |
mmAA |
sigAA |
scepsAA |
sccA |
| MSC |
|
nnAB |
mmAB |
sigAB |
scepsAB |
|
| MSC |
|
nnAC |
mmAC |
sigAC |
scepsAC |
|
| MSC |
k |
nnBB |
mmBB |
sigBB |
scepsBB |
sccB |
| MSC |
|
nnBC |
mmBC |
sigBC |
scepsBC |
|
| MSC |
l |
nnCC |
mmCC |
sigCC |
scepsCC |
sccC |
- MSORIG : specifies a particular tight-binding potential for silicon.
- MSTRANS : specifies an alternative tight-binding potential for silicon.
- MULLERBROWN : specifies the 2D Muller-Brown potential.
- MULTIPOT: Use the flexible multiple potential scheme to define your system.
Different atoms can interact via different types of potential and a particular atom can be associated
with more than one type of potential. Detailed instructions to use this scheme are given
as comments in multipot.f90, pairwise_potentials.f90 and isotropic_potentials.f90.
In theory, any subroutine with the correct signature can be used as a potential function. Only
the potentials in pairwise_potentials.f90 and isotropic_potentials.f90 have been tested.
At the time of writing, these are the LJ, WCA and harmonic-spring potentials. pairwise_potentials.f90
contains functions which are called for a pair of atoms at a time (e.g. harmonic springs). isotropic_potentials.f90
contains functions which require coordinates of all the atoms involved in the interaction. Although LJ and WCA are
pairwise potentials, it is more efficient to treat them as isotropic and loop over interacting pairs in the function
call rather than to call pairwise_lj for every pair of atoms (N2 function calls give a large performance overhead).
pairwise_potentials.f90 and isotropic_potentials.f90 contain generic functions for calculating the
first and second derivatives of isotropic pairwise potential functions. Detailed instructions are found in the source code.
An input file multipotconfig is used to specify which potential functions are to be used and
which atoms use each potential. For each type of potential in the system, multipotconfig should
contain the following:
- MULTIPULL npull: allow for multiple pulled pairs of atoms to be added through the PULL keyword.
npull is the number of pairs added, where a separate entry with PULL is needed for everyone. This keyword
has to be before the first entry for PULL, otherwise the arrays are not correctly used.
POT
POTTYPE n scale nparams
[params]
followed by a list of atom indices which will use this potential (the format of the list will depend
on the particular potential).
POTTYPE is a string identifier for the type of potential being used,
n is the number of atoms using this potential and scale is the energy unit for this potential
(which should be set to 1.0 for at least one potential). nparams is the number of potential-specific
parameters which are required, and [params] is a list of these parameters.
Be aware that using MULTIPOT will give a slight performance hit, because of the several function calls and bookkeeping that
is associated with each call to POTENTIAL. This scheme is intended for playing around with composite potentials, for systems which are small enough that performance is not a big issue, or for systems involving many different potential functions which will be much too fiddly to hard-code a single subroutine.
- MULTIPERM : instead of basin-hopping, systematically span the permutation space of the multiset containing particle labels. Uses a more general algorithm than ENPERMS .
- MULTIPLICITY xmul: specifies the multiplicity of the electronic state in DFTB
calculations.
- MWPOT: calls a Stillinger-Weber type potential with parameters appropriate for water[#!MolineroM09!#].
- NAPSHIFT version norm ε α fact Nin
Nhidden Nres: uses the NapShift ANN to incorporate chemical shift restraints during the optimization procedure. The string version specifies the method for combining the NapShift potential with the force field: MERGE computes the total energy as
(1 - α)VFF + αVNapShift, and NOMERGE computes the total energy as
VFF + αVNapShift. The string norm specifies how the NapShift energy is normalized: NORM divides the NapShift energy and forces by Nres, SQRTNORM divides the energy and forces by
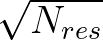 , and NONORM does not normalize the energy and forces. The value ε determines the width of the flat-bottomed NapShift potential. The value α is the weight of the NapShift energy and gradients as described previously. The string fact specifies the activation function of the NapShift ANN: either ELU or TANH. The integer Nin is the number of input nodes,
Nhidden is the number of hidden nodes, and Nres is the total number of residues of the protein. The chemical shifts must be specified in a chemshifts.dat file, the BLOSUM62 alignment must be specified in a blosum file, and the indices of the atoms making up the φ, ψ, χ1, and χ2 angles must be specified in a dihedral_indices file. Scripts for generating these files can be found in softwarewales/SCRIPTS/NAPSHIFT. The mean of each input value must be specified in an inputmean file and the weights of each NapShift ANN must be specified in a weights_ATYPE file, where ATYPE is one of C, CA, CB, H, HA, or N. The most recent versions of these files can be found in softwarewales/SCRIPTS/NAPSHIFT/weights. An example data file line could look like the following:
, and NONORM does not normalize the energy and forces. The value ε determines the width of the flat-bottomed NapShift potential. The value α is the weight of the NapShift energy and gradients as described previously. The string fact specifies the activation function of the NapShift ANN: either ELU or TANH. The integer Nin is the number of input nodes,
Nhidden is the number of hidden nodes, and Nres is the total number of residues of the protein. The chemical shifts must be specified in a chemshifts.dat file, the BLOSUM62 alignment must be specified in a blosum file, and the indices of the atoms making up the φ, ψ, χ1, and χ2 angles must be specified in a dihedral_indices file. Scripts for generating these files can be found in softwarewales/SCRIPTS/NAPSHIFT. The mean of each input value must be specified in an inputmean file and the weights of each NapShift ANN must be specified in a weights_ATYPE file, where ATYPE is one of C, CA, CB, H, HA, or N. The most recent versions of these files can be found in softwarewales/SCRIPTS/NAPSHIFT/weights. An example data file line could look like the following:
NAPSHIFT MERGE NORM 0.25 0.7 ELU 90 26 12
- NATB: specifies the sodium tight-binding potential of Calvo and Spiegelmann.
This potential can be guided by also specifying GUPTA 21 in the data file.
- NCAP: calls the capsid model [#!Wales05!#], consisting of pentagonal building blocks.
- NEON: introduces a diatomics-in-molecules calculation for
a neutral, cationic or electronically excited neon cluster. See also
GROUND, PLUS, TWOPLUS and STAR.
- NEWJUMP prob: for (serial)
`parallel' runs specifies a jump probability between runs
(parallel tempering) of prob.
See the BHPT keyword for a better alternative.
- NEWRESTART nrelax nhs MD newrestemp: reseed runs if the energy does not decrease within nrelax steps.
nhs is the number of hard sphere moves used to produce the new starting configuration.
If nhs=0 (the default) then the geometry is changed by reseeding. If MD is present, then a short AMBER or CHARMM MD run is performed at temperature newrestemp (specified in K, default 1000K) to generate the new configuration.
If the `F' argument appears for AVOID then
NEWRESTART will not reseed.
- NEWTSALLIS q: specifies that steps are accepted/rejected using Tsallis statistics with the
given value of q, rather than the usual Boltzmann condition. This version is slightly different from "TSALLIS" keyword.
- NOCHIRALCHECKS: disables checks for inversion of CA atoms and chiral side-chains for ILE and THR.
- NOCISTRANSCHECKS: disables checks for isomerisation of the peptide bond. This is equivalent to using the CISTRANS keyword.
- NOCISTRANS minomega: set on by default with a threshold minomega of 150 degrees.
If an amide bond is deformed to a angle below the specified threshold, the structure is discarded.
i.e. with | ω| <minomega. minomega defaults to 150 degrees.
For proline every ω is allowed. To enable cis-trans isomerisation, the CISTRANS keyword should be used.
- NOCISTRANSDNA minomega: should be specified when working with DNA in AMBER to ensure the correct bonds are
checked. As above, the deformation threshold minomega can be set. It is defaulted to 150 degrees.
- NOCISTRANSRNA minomega: should be specified when working with RNA in AMBER to ensure the correct bonds are
checked. As above, the deformation threshold minomega can be set. It is defaulted to 150 degrees.
- NOFREEZE: don't freeze the core atoms when doing the initial geometry optimisations in
a run where SEED is specified.
- NOGETROT: do not include the rotational partition function; for use with
FEBH and CNBH.
- NOGETVIB: do not include the vibrational partition function; for use with
FEBH and CNBH.
- NOGETTRANS: do not include the n3/2 factor in the translational part of the partition function; for use with
FEBH and CNBH.
- NOGETSYM: do not include the factor
n!/oα in the partition function for use with
FEBH and CNBH.
- NOINVERSION: turns off inversion of structures for distance
metric in minpermdist.
- NOPHIPSI: used with the CHARMM keyword to specify twisting of
sidechain dihedrals only.
- NOPOINTS: if USEMINDATA is specified then a points.min file is created
with entries corresponding to min.data unless NOPOINTS is set. The points file is
not used for identifying minima; instead we check the energy and the principal moments of
inertia.
- NOREGBIAS: see MLPVB3.
- NORESET: by default the configuration point is set to that of the
quench minimum in the Markov chain during a basin-hopping simulation. This
keyword turns off the resetting so that the geometry varies continuously.
- NOTE : the rest of the line is ignored.
- NPAH n: calls a finite system of polycyclic aromatic hydrocarbons (PAH) interacting via
Williams' potential [#!Williams80!#]. The PAH ID n defines the PAH molecule: 1 for coronene, 2 for
pyrene, 3 for benzene.
- NRELAXRIGID MinR MinA: Minimizations will use rigid body coordinates for MinR steps and then atomistic coordinates for MinA steps. This is to be used with RIGIDINIT keyword.
- NTIP n: calls a member of TIP family of potentials for water molecules within a rigid-body
framework.
1≤n≤5. n = 1: TIPS water; n = 2: TIPS2 water; n = 3: TIP3P water;
n = 4: TIP4P water; and n = 5: TIP5P water.
- ODIHE : order parameter—requires documentation.
- OEINT : interaction energy between 2 peptides will be used as an order parameter—requires
further documentation.
- OHCELL : allow point group operations for a cubic
supercell in subroutine minpermdist.f90 permutational distance minimisation.
- OPEPHIRE option: alternative use OPEP or HIRE, specifies the use of the HiRE and OPEP potentials, option is Protein, RNA, DNA, MixedRNA or MixedDNA. The following additional files are needed for OPEP protein simulations: conf_initiale.pdb, ichain.dat, scale.dat, cutoffs.dat, parametres.top and parametres.list. For RNA and DNA simulations the following files are required: conf_initiale.pdb, conf_initiale_RNA.pdb, ichain.dat, scale_RNA.dat, cutoffs_RNA.dat, parametres_RNA.top, baselist.dat, bblist.dat, chargeatm_RNA.dat and protonated.dat.
- OPP mode rcut xplor k φ params k_lower k_upper
specifies that the Oscillating Pair Potential potential should be used. This potential can
have several wells, depending on the value of k, a frequency parameter, and φ, a phase parameter.
The XPLOR smoothing function is used to make the potential and its first derivatives go smoothly to zero at rcut, with
the smoothing beginning at xplor (which must be less than rcut).
mode can be either 0, 1, 2 or 3. mode 0 specifies the normal potential, and can be either a cluster or with periodic
boundary conditions with an orthogonal unit cell using the PERIODIC keyword. mode 1 or 2 requires periodic boundary conditions.
The PERIODIC keyword must be used, but the unit cell parameters from it will be ignored. Instead, the second last line of the coords file
will be interpreted as unit cell angles and the last line as unit cell lengths for a triclinic cell and will be optimised. Since there
are combinations of unit cell angles that are physically impossible, steps will be rejected if such combinations are detected. In mode 2, as
well as the cell being optimised, potential parameters specified by params (an integer between 1 and 3)
will also be optimised. If the lowest bit of params is set, k will be optimised, and if the second bit is set, φ will be optimised.
In mode 3, potential parameters will be optimised as for mode 2, but there are no periodic boundary conditions.
For mode 2 and 3, the lower and upper bounds for k can be specified with k_lower and k_upper.
If k steps outside these bounds, a harmonic repulsion will be turned on to push k back towards the range.
- ORBITALS : orbital localisation rotation potential.
- ORCA charge spin path : call ORCA as a subprocess to calculate the potential.
charge specfies the charge of the system and spin specifies the spin multiplicity.
path is the path to the ORCA executable, which must be present on the system, default
`orca' for when the executable is already in the PATH. Note that the full path to ORCA must be
specified if parallelisation is to be used within the ORCA subprocess. This keyword automatically turns
on READZSYM, so an additional input file called ZSYM must be present in the
working directory that specifies the identities of the atoms. There must also be present
a file called ORCA.template, which is the ORCA input file containing everything
except the atomic coordinates. The ORCA run must produce a .engrad file that
contains the energy and gradients at the location specified by GMIN, so should probably
contain the ENGRAD keyword. For example, to
run a DFT calculation for H2, the contents of ORCA.template could be
! B3LYP ENGRAD def2-TZVP
and the contents of ZSYM should be
H H
The starting coordinates should be specified in coords as normal.
Additional entries in the ORCA.template file are probably desirable. The following
example, for a Restricted Hartree-Fock calculation with a small basis set, reduces the
amount printed in the ORCA.out file, since that file is not used by GMIN. It
disables calculation of the molecular dipole moment, which ORCA calculates by default but
is not required. The %PAL line requests ORCA use MPI with 16 processes for the
calculation, for which an appropriate ORCA executable must be specified.
! RHF ENGRAD def2-SVP MiniPrint
%PAL NPROCS 16 END
%elprop
Dipole False
end
- ORGYR : radius of gyration will be calculated as an order parameter—requires
further documentation.
- OSASA : order parameter—requires documentation.
- P46: specifies a 46-bead three-colour model polypeptide.
See also the BLN keyword, which implements this potential in a more
general way and uses unit bond lengths.
- PACHECO: specifies the intermolecular Pacheco-Ramelho potential for C60.
The Axilrod-Teller contribution, specified with the AXTELL keyword, is included
when the RMS force falls below the value entered with GUIDECUT.
- PAH: specifies a polycyclic aromatic hydrocarbon potential.
- PAHA n: calls a finite system of polycyclic aromatic hydrocarbons (PAH) interacting
via a general anisotropic potential developed from first principles [#!TottonMK10!#]. The PAH ID
n defines the PAH molecule: 1 for benzene, 2 for naphthalene, 3 for anthracene, and 4 for
pyrene.
- PAHAGENRIGID n: calls a finite system of polycyclic aromatic hydrocarbons (PAH) interacting
via a general anisotropic potential developed from first principles [#!TottonMK10!#]. This potential has
been adapted to the GENRIGID framework for rigid bodies (also use the RIGIDINIT keyword). The PAH ID
n defines the PAH molecule: 1 for benzene, 2 for napthalene, 3 for anthracene, and 4 for pyrene.
- PAIRDIST pair1a pair1b pair2a pair2b...: enables tracking of the distances between pairs of atoms during a GMIN run. Atom pairs may
be specified in the keyword definition as shown, or in the file pairdist with a pair of atoms on each line. The pair distances are
calculated after each quench and printed in pairdists.
- PAP npatch alpha s cosdel epsilon: specifies a patch anti-patch potential.
Each body consists of a Lennard-Jones core, range parameter alpha,
with npatch patches and npatch anti-patches. The patch anti-patch
attraction has a range specified by s, a width specified by cosdel
and a strength specified by epsilon. If npatch is set to zero,
site information will be read in from the file papsites.xyz.
- PARALLEL npar GMIN: npar is the number of parallel runs within GMIN.
If the word "GMIN" is present as the second argument, the calculation will proceed until the
same lowest energy is recorded in each trajectory.
- PBGLUE: specifies a glue potential for lead.
- PERCOLATE dist comp cutoff: specifies that particles are prevented from evaporating using a system based on maintaining a percolating graph of particles, rather than using a container, with dist as the maximum distance at which particles are considered to be connected. An optional harmonic compression potential can be specified with force constant comp, to be turned off below RMS force cutoff. Currently, the step size is limited to dist squared.
- PERIODIC boxlx boxly boxlz alpha beta gamma: specifies periodic boundary conditions for
potentials which understand such a directive. The three variables boxlx, boxly, boxlz are the
box lengths. If only one box length is given, the others are set to the same value to give a cube.
The three variables alpha, beta, gamma are the box angles in radians. If the box angles are
not given, the box is taken to be orthorhombic.
- PERMDIST x: minimise distances between
the coordinates in files coords and the
fixed coordinates in file finish with respect to permutational isomerisation.
Requires the auxiliary file perm.allow to specify permutable atoms, otherwise
all atoms are assumed to be permutable. The absence of a perm.allow
file is considered a mistake for CHARMM runs.
The parameter x is the distance tolerance for assigning atoms to orbits
in the myorient standard orientation routine, default value 0.001.
If x is too small it is possible for permutational isomers to be missed,
however, if x is too large then runs with the AVOID keyword can
slow down by an order of magnitude.
The first line of the perm.allow file must contain an integer
that specifies the number of primary groups of interchangeable atoms.
The groups then follow, each one introduced by a line with two integers p and s
that specify the number of permutable atoms in the primary group and the number of other sets
of permutable atoms associated with the primary set.
s may be zero.
Each secondary set of permutable atoms has p members.
The following line contains the indices of the p permutable atoms
in the primary set and then
the indices of the atoms in each of the s secondary sets, one set at
a time.
For the phenylalanine example illustrated we must allow three other pairs of
atoms to exchange if we swap 7 and 8. Hence a suitable perm.allow entry is
1
2 3
7 8 5 6 1 2 3 4
Here n = 2 and s = 3: if we exchange 7 and 8 then we must also exchange 5 and 6,
1 and 2, and 3 and 4. There are two atoms in each of the three secondary sets,
since we have specified 7 and 8 as the two primary atoms.
Here is an example perm.allow file for a water trimer using
the flexible QTIP4PF potential, where the energy is invariant to permutations
of water molecules and to exchanges of hydrogens in the same molecule. However,
hydrogens cannot exchange between different oxygens:
4
3 2
1 4 7 2 3 5 6 8 9
2 0
2 3
2 0
5 6
2 0
8 9
The first group of three oxygens has two atoms that must move with each oxygen,
i.e. atoms 2 and 3 for oxygen 1, etc. Hydrogen permutations for each oxygen are
allowed by the three following groups. This scheme allows atoms to appear in more
than one group. There must be a group containing each complete set of permutations
in order for permutation-inversion isomers to be recognised. The format
is compatible with an older scheme, where only pair swaps were allowed for
associated atoms, but now allows for more general permutations.
Scripts to generate allowed permutations automatically for CHARMM and AMBER are available from
the group web site. It is essential to use symmetrised versions of the corresponding
force fields!
- PERMINVOPT x: minimise the distance between the geometries in
coords and finish with respect to permutations, as well
as overall translation, rotation, and inversion. See
PERMOPT for distance optimisation without the inversion.
To specify specific groups of
permutable atoms a perm.allow file is needed, unless all atoms (or rigid bodies)
are considered permutable.
For rigid bodies the file rbsymops can be used to specify symmetry
operations of each molecule, and the distance will then also be
minimised with respect to internal symmetry operations.
To minimise the Euclidean distance between two configurations with no permutations
a perm.allow file with the single entry `0' can be used.
The configurations are then aligned as if they were rigid bodies decorated
with fixed sites.
The parameter x is the distance tolerance for assigning atoms to orbits
in the myorient standard orientation routine, default value 0.001.
If x is too small it is possible for permutational isomers to be missed,
however, if x is too large then runs with the AVOID keyword can
slow down by an order of magnitude.
- PERMOPT x: minimise the distance between the geometries in
coords and finish with respect to permutations, as well
as overall translation and rotation, but not the inversion. To specify specific groups of
permutable atoms a perm.allow file is needed, unless all atoms (or rigid bodies)
are considered permutable.
For rigid bodies the file rbsymops can be used to specify symmetry
operations of each molecule, and the distance will then also be
minimised with respect to internal symmetry operations.
To minimise the Euclidean distance between two configurations with no permutations
a perm.allow file with the single entry `0' can be used.
The configurations are then aligned as if they were rigid bodies decorated
with fixed sites.
The parameter x is the distance tolerance for assigning atoms to orbits
in the myorient standard orientation routine, default value 0.001.
If x is too small it is possible for permutational isomers to be missed,
however, if x is too large then runs with the AVOID keyword can
slow down by an order of magnitude.
- PLANCK h: specifies the value of Planck's constant.
This is used in calculating partition functions in FEBH and CNBH.
SI units are needed for TIP4PGR and AMBER12.
A value of the Planck constant in reduced LJ usits for a specific system is
needed for intert gases with LJ.
- PLUS: when combined with keywords NEON or ARGON
uses a diatomics-in-molecules potential for the singly charged cation.
- POLYHEDRA overlap compress the system is composed of convex polyhedra, with a harmonic
repulsion of force constant overlap dependent on the distance by which a pair of polyhedra
overlap with each other, and a harmonic attraction of force constant compress to bring
the particles towards the origin. All polyhedra in the system are identical and their vertices
are specified in the file polyhedron_vertices.dat. The first line of this file is
the number of vertices, which must be at least 4, and the following lines give the
coordinates of each vertex.
- POWER ipow: ipow is the initial power for shifts in the old line minimisation routine
for conjugate gradient. LBFGS minimisation should be used instead.
- POWERLAW a b: specifies a power law potential of the form
for a and b positive real numbers, so that the first term is attractive and the second repulsive.
Typical values are a = 2 and b = 0.5. See arXiv:2403.00594, arxiv:1405.5428 and arxiv:1612.09233.
- PROJI: turns on projection operator to enforce I point group symmetry
in mylbfgs.f. The geometry is projected after every proposed step.
- PROJIH: turns on projection operator to enforce Ih point group symmetry
in mylbfgs.f. The geometry is projected after every proposed step.
- PRTFRQ n: prints the energy every n steps; default is every step.
Should now work for basin-hopping, basin-sampling and parallel tempering runs.
- PRINT_PTGRP tol1 tol2 tol3: Print point-group classification and order for each structure saved in the file lowest. The three tolerances are optional: tol1 is a dimensionless tolerance for diagnosing degeneracies in (normalised!) principal moments of inertia, tol2 is the distance tolerance used for detecting point-group symmetry operations, and tol3 is a dimensionless tolerance used for testing whether a generated rotation matrix (representing a symmetry operation) is new. The default values are 0.001, 0.2 and 0.1, respectively.
- PRINT_MINDATA eigen_tol: Print the energy, log product of (non-zero) Hessian eigenvalues, and the order of point group for each minimum in the file lowest, instead of the usual comment line. The optional real-valued parameter eigen_tol sets the tolerance for diagnosing negative (less than -abs(eigen_tol)) or zero Hessian eigenvalues. The default value is eigen_tol = 0.001. Note that if a negative eigenvalue is diagnosed or if the number of zero eigenvalues is unexpected, then the ``minimum'' will not be written to the file lowest.
- PTMC histmin histmax ptemin ptemax pttmin pttmax exchprob nequil ptsteps nenrper hbins:
requests a standard parallel tempering MC run.
This keyword also specifies the energy range for the histogram of quench energies,
histmin to histmax,
the energy range for the histogram of instantaneous configurations, ptemin to ptemax,
the temperature range (pttmin and pttmax),
the probability of attempting an exchange exchprob, the
number of equilibration steps, nequil,
the number of parallel tempering MC steps without quenching, ptsteps,
the number of bins for the histogram of instantaneous potential energy, nenrper, and
the number of bins for the histogram of quench energies, hbins.
Should be used together with the MPI keyword. (This option is only available if the source is compiled with an MPI enabled.)
- PTSTST npatch eps cosdel kappa Specifies a patchy site-to-site potential,
similar tp PAP but with Gaussian patches on the surface of the particle. The
number of patches is determined by npatch. eps is the strength of the
Gaussian patch attraction, cosdel is the effective range of the Gaussian and
kappa is the Yukawa parameter for the core repulsion (currently unused as the
core is Lennard-Jones rather than Yukawa).
- PULL a1 a2 f: apply a static force to the potential, equivalent to adding
the term
Vpull = - f (za1 - za2). Here za1 and za2 are the z
coordinates for atoms a1 and a2, and f specifies the force.
This potential is designed to simulate a pulling experiment with static force where
a molecule is pulled along the z axis from atoms a1 and a2.
- PY sig0 eps0 [cutoff XYZ xbox ybox zbox]: specifies a rigid-body multisite Paramonov-Yaliraki[#!ParamonovY05!#] potential with parameters σ0 and
ε0. Optional arguments are
a cutoff distance in absolute units, a specification of which directions have periodic boundary
conditions, and the size of the periodic box in units of the cutoff. The parameters for and setup
of the rigid body, even if it is just one site, must be specified in a file pysites.xyz.
There are two formats for pysites.xyz: identical particles and n-ary mixtures. For identical
particles, the format is:
[number of sites in molecule]
[blank line]
site [x position] [y] [z] shapes [rep coeff] [att coeff] [a11] [a12] [a13] [a21] [a22] a[23] orient [p_x] [p_y] [p_z]
...
site [x position] [y] [z] shapes [rep coeff] [att coeff] [a11] [a12] [a13] [a21] [a22] a[23] orient [p_x] [p_y] [p_z]
For an n-ary mixture,
0 [the number zero]
[arity]
[blank line]
[number fraction of this type of molecule]
[number of sites in molecule]
[normal site lines]
...
[blank line]
[second number fraction]
[number of sites in second type]
[normal site lines]
...
For example, for a single site molecule,
1
site 0.0 0.0 0.0 shapes 1.0 1.0 0.5 0.5 0.2 0.5 0.5 0.2 orient 0.0 0.0 0.0
Or, for example, for two-to-one mixture of single sites and pairs,
0
2
0.66
1
site 0.0 0.0 0.0 shapes 1.0 1.0 0.5 0.5 0.2 0.5 0.5 0.2 orient 0.0 0.0 0.0
0.34
2
site 0.0 0.5 0.0 shapes 1.0 1.0 0.5 0.5 0.2 0.5 0.5 0.2 orient 0.0 0.2 0.0
site 0.0 -0.5 0.0 shapes 1.0 1.0 0.5 0.5 0.2 0.5 0.5 0.2 orient 0.0 -0.2 0.0
- QALCS qalcsmode cutoff: perform Quench-Assisted Local Combinatorial Search to locate a biminimum for a multicomponent system. (More general than HOMOREF, which works only for binary systems.) The search comprises a sequence of quench-assisted atom swaps, where each step involves a sweep through the local neighbourhood structure induced by the ``interchange'' distance metric. There are many ways of doing this: qalcsmode=0 specifies a slow steepest-descent-like search in permutation space; whereas modes 1 through 5 specify alternative schemes that are more efficient for larger systems. Modes 4 and 5 admit an additional (optional) integer parameter cutoff, which specifies that only the first cutoff entries in the sorted local neighbourhood are to be considered. The sorting is done by approximate swap gain.
- QALCS_SURF: specifies that QALCS should include surface vacancies as a separate species, akin to dynamic-lattice-search-type methods but relies on a different construction of surface vacancies. The scheme constitutes a deterministic way of refining the surface of a cluster within the QALCS framework, and it can be used with or without the
QALCS keyword (for single- or multi-component clusters). Note that during the surface optimisation all particles are treated as being the same.
- QC1 n: Perform a standard quantum computing calculation with n number of qubits. The following additional input files are required:
- qc_circuit.dat: A file containing the quantum circuit to be executed. The number of columns should equal n nqubits, with the first column representing qubit 0, the second column representing qubit 1, and so on.
Each row of the file should represent a single time step in the quantum circuit, with the entries in each row specifying the gates to be applied to each qubit at that time step. A three-letter code should be used to indicate the type of gates:
- IDE: Identity gate
- HAD: Hadamard gate
- PAX: Pauli-X gate
- PAY: Pauli-Y gate
- PAZ: Pauli-Z gate
- RXF: Fixed parameterised rotation gate around the X-axis (Angle specified with qc_fixed_rotations.dat)
- RYF: Fixed parameterised rotation gate around the Y-axis (Angle specified with qc_fixed_rotations.dat)
- RZF: Fixed parameterised rotation gate around the Z-axis (Angle specified with qc_fixed_rotations.dat)
- RXP: Parameterised rotation gate around the X-axis (Angle specified with coords)
- RYP: Parameterised rotation gate around the Y-axis (Angle specified with coords)
- RZP: Parameterised rotation gate around the Z-axis (Angle specified with coords)
- CXC CXT: Controlled-X gate for control qubit and target qubit respectively (Note: If there is a gap between control and target qubits, use IDN to indicate that the intermediate qubits are not involved in the gate)
- CZC CZT: Controlled-Z gate for virtual control qubit and target qubit respectively (Note: If there is a gap between control and target qubits, use IDN to indicate that the intermediate qubits are not involved in the gate)
- SWC SWT: SWAP gate for virtual control qubit and target qubit respectively (Note: If there is a gap between control and target qubits, use IDN to indicate that the intermediate qubits are not involved in the gate)
- LIN: Linear chain of CNOT gates, with the control qubit on the first column specified (do not skip qubits)
- LID: Linear chain of CNOT gates, with the target qubit on the first column specified (do not skip qubits)
- ZZC ZZT: Parameterised rotation-ZZ gate for Control qubit and target qubit respectively, angle specified with coords (Note: If there is a gap between control and target qubits, use IDN to indicate that the intermediate qubits are not involved in the gate)
- qc_coeffs.dat: A file containing the real coefficients for the Hamiltonian being simulated (should be same length as qc_operators.dat).
- qc_fixed_rotations.dat: A file containing the angles for the fixed parameterised rotation gates (should be same length as number of fixed parameterised rotation gates in qc_circuit.dat). Default should be a value of 0.0.
- qc_operators.dat: A file containing the operators for the Hamiltonian being simulated in terms of Pauli strings (should be same length as qc_coeffs.dat).
- QCPERIODIC: Works alongside QC1, specifies that the output angles should be between -π and π.
- QCUCC: Works alongside QC1, specifies that unitary coupled cluster (UCC) ansätze are simulated instead of gate-based quantum computation. The input files are different from standard QC1:
- qc_ucc_hamiltonian.dat: A file containing the Hamiltonian for UCC simulation. Note for ease of simulation, Pauli strings are not used, and instead the Hamiltonian is specified by floats (in conjunction with qc_ucc_index.dat).
- qc_ucc_index.dat: A file containing the indices of the Hamiltonian in qc_ucc_hamiltonian.dat, where the first integer given per line is the row index and the second integer is the column index. The length of this file should be the same as qc_ucc_hamiltonian.dat.
- qc_ucc_initial.dat: A file containing the initial state to initialise the UCC simulation (in binary).
- qc_ucc.dat: A file containing the excitation operators for UCC ansätze. There should be a maximum of four columns, with the single excitations
a†iaj given as (i, j, 0, 0) and the double excitations
a†ia†jakal given as (i, j, k, l).
The files qc_circuit.dat, qc_coeffs.dat, qc_fixed_rotations.dat and qc_operators.dat are not used for UCC simulations.
- QCORDER: Works alongside QC1, specifies the binding of the same operators to different parameterised gates (RXP, RYP, RZP, ZZC/ZZT).
The length of the file should be the total number of parameterised gates in the circuit, and the maximum integer in the file should be the number of parameters in coords.
If QCUCC is used, indicate this with the additional file qc_ucc_order.dat. If not, use qc_order.dat.
- QCSCALE: Works alongside QC1, specifies how each parameterised gate (RXP, RYP, RZP, ZZC/ZZT) is scaled with the corresponding scaling factors are specified.
The length of the file should be the total number of parameterised gates in the circuit.
If QCUCC is used, indicate this with the additional file qc_ucc_scale.dat. If not, use qc_scale.dat.
- QCMULT: Works alongside QC1 and QCUCC, specifies that an initial state with multiple computational basis states is used.
The file qc_ucc_initial.dat should contain the binary representation of each computational basis state, with the number of lines in the file corresponding to the number of computational basis states.
An additional file qc_ucc_initial_coeffs.dat should list the real amplitudes of each computational basis state.
- QCQML: Works alongside QC1, specifies that a quantum machine learning calculation occurs. By default, a classification task is carried out. The following input files should be included:
- qc_circuit.dat: In addition to the same lettering for standard QC1, the additional three-letter codes can be specified:
- RXI: Input rotation-gate around the X-axis, with angles specified by qc_input.dat.
- RYI: Input rotation-gate around the Y-axis, with angles specified by qc_input.dat.
- RZI: Input rotation-gate around the Z-axis, with angles specified by qc_input.dat.
- ZZI ZZT: Input rotation-ZZ gate for control and target qubits respectively, with angles specified by qc_input.dat.
- qc_input.dat: Specifies the input files for the datapoints used in the QML classifier in float format.
The number of rows indicates the number of datapoints, and the number of columns specifies the number of input values (should be the same as the total number of RXI, RYI, RZI and ZZI gates).
- qc_classes.dat: Specifies the classes of each datapoint in integer format. The number of lines should be the same as the number of datapoints.
The files qc_coeffs.dat and qc_operators.dat are not used in QCQML.
- QMLINIT: Works alongside QC1 and QCQML. Instead of using qc_circuit.dat and qc_input.dat, use this keyword to specify loading the initial states of each datapoint used.
The following files should be modified:
- qc_initial_index.dat: Specifies the index of the datapoint and the computational basis state in integer order. The first column's maximum value should equal the number of the datapoints.
The second column should have a maximum value of 2n. The coefficients of the initial state should be specified with qc_initial.dat.
- qc_initial_real.dat: Specifies the real coefficients of the initial state of all datapoints. The length of this file should be the same as qc_initial_index.dat.
- qc_initial_imag.dat: Specifies the imaginary coefficients of the initial state of all datapoints. The length of this file should be the same as qc_initial_index.dat.
- qc_input.dat: If no RXI, RYI, RZI or ZZI gates are used, specify a value of 0.0 for each datapoint used.
- QMLREG: Works alongside QC1 and QCQML, specifies that a regression task is carried out instead of classification. The following additional input file should be included:
- qc_coeffs.dat: A file containing the real coefficients for the Hamiltonian being simulated (should be same length as qc_operators.dat).
- qc_operators.dat: A file containing the operators for the Hamiltonian being simulated in terms of Pauli strings (should be same length as qc_coeffs.dat).
- qc_values.dat: A file containing the real values for each datapoint to fit towards. The number of lines should be the same as the number of datapoints.
- QMAX cgmax: cgmax is the tolerance for the
RMS force in the final set of quenches that are used to produce
the output for file lowest. The default is
cgmax= 10-3, but the appropriate value depends upon the system in question.
TIGHTCONV can be used instead.
- QUAD: requires documentation.
- QUCENTRE: sets the centre of coordinates to the origin (0,0,0) before each MC step is taken (so after each quench), but not during the minimisation itself unlike CENTRE.
- QUIPZ z: defines the atomic number z for the quip interface.
- QUIPLATT matrix: defines the components of a 3×3 lattice for the quip interface.
- QUIP_PARAM_FILE qpf: sets the quip parameters file. qpf is a string and the
default is quip_params.xml.
- QUIP_INIT_ARGS_STR ias: sets the quip parameter `init_args_str'.
ias is a string with default value `xml_label=default'.
- RADIUS radius: sets the radius of the container that prevents particles
evaporating during quenches. If unset the program calculates an appropriate value
based upon the volume per particle for close-packed material and the known pair
equilibrium distance for the given potential. The formula employed is
where n is the number of atoms and re is the pair equilibrium
separation.[#!kittel76!#] The `1' in this formula is to allow some extra space for
more open structures.
- RANDOMSEED: specifies that the random number generator should be seeded with system time after each quench, allowing simple parallel use. Currently functional only for the CHARMM and AMBER potentials.
- RANSEED i: integer seed for the random number generator. The number actually used is mod(i,10000)+1.
- RANDMULTIPERM n: randomly permute atomic labels every n basin-hopping steps. Intended for use in conjunction with keyword QALCS.
- RATESTOFREE states kT h/kT : reads a kinetic transition matrix from file
ratematrix for states nodes and performs a least squares fit to produce
relative free energies of the states. Files are dumped for the corresponding minima
of the least squares fit in min.data.n and for transition states in ts.data.n.
The transition states free energies are obtained from the connected minima and the
corresponding rates, with a conversion factor kT for the factor of kT that
multiplies the log ratio of forward and backward rates in the fit. Setting this conversion
factor to one will give dimensionless relative free energies in units of kT.
h/kT is a conversion factor that
makes the product with a rate dimensionless, e.g. picoseconds if the rates are per picosecond.
It is used to make the free energies for the transition states that are written to the new
ts.data file. The output files are appended with a number for the local minimum that
they correspond to.
- RATIO stepratio tempratio: adjusts stepsize and temperature independently.
stepratio is the target fraction of steps that move into a different well. Identity of
structures is determined by structural alignment; as such, the PERMDIST keyword and
appropriate auxiliary files are required. tempratio is the target acceptance ratio for
those steps that move into new basins. If a negative number is supplied for either, the
ratios is printed, but the corresponding parameter is not adjusted.
- RBSYM: specifies that internal symmetry operations permuting equivalent sites for
rigid-body building blocks exist. The operations are read in from a file rbsymops, which must
exist in the working directory. The first line of the file is an integer that specifies the number
of operations generated by proper axes of rotation including the identity operation; the operations
are then read in one at a time in a representation consisting of a unit vector, defining the
axis in the reference frame, and an angle in degrees, describing the magnitude of rotation about
that axis. The first operation has to correspond to the identity operation.
This keyword has to be combined with PERMDIST so that structures are aligned and
a distance metric considered based on permutations of identical rigid
bodies and any internal symmetry operation of each rigid body that is a symmetry
of the potential energy function.
- READZSYM: reads element symbols from file ZSYM in free format.
Having the correct symbols is required for XTB jobs unless there is only one atom type.
- REAXFF: specifies the use of the REAXFF force field. The following additional files are needed:
fort.3 or start, which contain the starting geometry, fort.4, which contains
a specific force field parametrization, control, which contains additional force field settings.
We recommend using ReaxFF with a start file for the coordinates, in which case fort.3 will only
be used to read atomic symbols and number of atoms in the system. The format of fort.3 is BGF and is format-sensitive.
- REAXFFDECOMP: print energy decomposition file (fort.200) for REAXFF.
- RELAXFINALQUENCH: This means final quench is done in atom coordinates. Useful if you want to compare final energies. This is to be used with RIGIDINIT keyword.
- RESERVOIR n: for use in combination with PTMC. A reservoir of local minima
is used for the lowest temperature replicas up to cpu number n. The default is
n = 0, which means that only the lowest replica used the reservoir. Visit statistics are
dumped for replicas that use the reservoir, but exchanges only occur when the lowest
non-reservoir replica inherits a configuration. This keyword requires files
min.data and points.min in the current working directory in PATHSAMPLE
format. See also EXEQ.
- RESIZE resize: all the coordinates are multiplied by resize after
they have been read in, before any other operations are performed. This command is useful
for scaling results obtained with one potential for a system with a different pair
equilibrium distance.
- RESTART nrelax nhs: reseed runs if a step in not accepted
within twice nrelax steps.
nhs is the number of hard sphere moves used to produce the new starting configuration.
If nhs=0 (the default) then the geometry is changed by reseeding.
- RESTORE dumpfile intEdumpfile: restore a previous GMIN run from a dumpfile.
The number of basin-hopping steps performed will be the number of steps specified with the keyword STEPS,
minus the number that were completed
at the point the dumpfile was created. This option is not available before version 2.3.
If you are using the A9INTE keyword, you can specify the interaction enthalpy
dump file to restore from as a second argument.
- RGCL2: specifies a DIM rare gas-Cl2 potential.
- RIGIDINIT: This will turn on the local rigid body framework. This keyword needs two additional input files: coordsinirigid and rbodyconfig. coordsinirigid should contain the coordinates of all the atoms whether they are part of local rigid bodies or not. During initialization, GMIN will select appropriate coordinates and throw away irrelevant ones. The format is:
x1 y1 z1
x2 y2 z2
...
...
xn yn zn
The file rbodyconfig defines the rigid bodies, with the following format:
GROUP NoAtomsInGroup
Atom 1
Atom 2
...
...
Atom N
The keywords RELAXFINALQUENCH and NRELAXRIGID can be used in conjuction with RIGIDINIT.
- RINGROTSCALE factor: when applying cartesian moves with CHARMM, amino acid rings are moved as rigid units. Setting factor (default 0.0) between 0.0 and 1.0 will apply a random rotation to these rings during step taking. The suggested value is 0.1 to prevent the regular formation of high energy structures.
- RKMIN: specifies a Runga-Kutta minimisation scheme.
Very inefficient.
- RMS rmslimit rmstol rmssave lca best: used with CHARMM keyword to
specify that the RMSD compared to a reference geometry is calculated. The reference geometry must
be given in xyz-format in an additional file compare. rmssave is an integer
that specifies the number of lowest energy geometries and RMSD ≤ rmslimit
to save. Geometries are only saved if their RMSD's are more than rmstol
different. The flag lca controls whether the all-atom RMSD (lca=0) or the
Cα-RMSD
(lca=1) is calculated. The flag best determines which structure is compared to the reference
after each quench. best=0 implies the current quench minimum and best=1 implies the current best (lowest energy) minimum. If TRACKDATA is also specified, the RMSD calculated after each quench is produced in the file `rmsd' in gnuplot readable format.
- ROTAMER maxchange pselect occuw (centre cutoff): Used with AMBER9 only. Specifies that rotamer moves should be taken. Every step, up to maxchange rotamers may be selected with a probability pselect. occuw determines the minimum % occupation a rotamer must have to be selected from the library[#!lovelljm00!#] when making a change. For example, occuw = 0.004 restricts possible rotamer choice to those with a greater than 0.4% occupation. If you want to focus rotamer changes around a ligand/binding pocket, the optional centre and cutoff arguements may be used. centre specifies the residue of interest (for example a ligand), and cutoff the limiting distance from this centre that rotamers may be changed. The selection probability decreases linearlly from the centre residue. To use these moves, you need three files found in the SCRIPTS directory: PdbRotamerSearch, penultimate.lib and rotamermove.csh in your working directory for each run.
- ROTAMERMOVE frequency maxrotmoves selectionscheme: Specifies that rotamer moves should be taken using the sequence-dependent rotamer library. Rotamer moves are made every frequency steps and a random number of side chains up to maxrotmoves are changed. selectionscheme specifies how the amino acids are selected for rotamer moves, currently ``UNIFORM'' is the only option. Rotamers are chosen for a particular amino acid based on its adjacent amino acids and its predicted occupation probability.
- ROTATEHINGE freq factor: For use with the plate-folding potential (but could possibly be used elsewhere as well). An input file hingeconfig is used to define hinges between sets of rigid bodies. Every freq steps, the set of rigid bodies on the ``moving side'' of the hinge are rotated about the hinge axis by a random angle between
- π×factor and
π×factor.
For each hinge, hingeconfig contains the following three lines:
nmove, the number of plates on the ``moving side'' of the hinge
[plate_indices] , a list of rigid body indices for the moving plates
atom1 atom2 atom3 atom4.
The hinge axis connects the midpoints of the lines connecting the two pairs (atom1, atom2) and (atom3, atom4). For the plate potential, each of these atom pairs should straddle the hinge and be connected by a stiff spring. atom1 lies on the same plate as atom3. atom2 and atom4 are on the opposite side of the hinge.
- ROTATERIGID freq factor: for use with the generalised rigid body framework,
RIGIDINIT. Randomly rotates each rigid body in the system by a factor of 2π multiplied by factor every freq steps.
- ROTATERIGIDAXIS freq factor fixed: for use with the generalised rigid body framework,
RIGIDINIT. Randomly rotates each rigid body in the system around the principal axis of rotation, by a factor of 2π multiplied by factor every freq steps. If rotation by a specific angle is required, mention the angle in radians at the fixed placeholder, otherwise leave it blank. (For example, fixed = 1.57 leads to a rotation by 90 degrees.)
- SANDBOX: Specifies that the sandbox potential should be used. This keyword requires an auxiliary input file rbdata and produces an auxiliary output visualization file sandout.xyz. See the Sandbox section of this documentation for information on producing rbdata files and for examples.
- SAVE nsave: nsave is an integer that specifies the number
of lowest
energy geometries to save and summarise in the file lowest.
Arrays are now dynamically allocated, so any positive integer can be specified.
Note that if nsave is large, minima that are not in the Markov chain
might also be written out which might be high energy non-physical
conformations.
- SAVEFRQS: save all normal mode frequencies for use with CNBH and FEBH if a quantum vibrational
partition function is required. Creates direct access file min.frqs.
- SAVEMULTIMINONLY: specifies that only multiminima are to be considered for the final dump to lowest.
- SAVEINTE nsaveinte: nsaveinte is an integer that specifies the number of lowest
interaction enthalpy geometries to save and summarise in the file intelowest. See A9INTE.
- SEMIGRAND_MU mu_1 ...mu_M: specifies the semi-grand chemical potentials, relative to the first species, for use in conjunction with the keyword LFLIPS. The number of values ought to match the number of different species minus one (i.e. M=NSPECIES-1).
- SETCENTRE x y z: Sets the centre of mass/coordinates (before the initial quench) to (x,y,z). For example, SETCENTRE 0.0 0.0 0.0
would translate the centre of mass to the origin.
- SETCHIRAL: For use with AMBER9, NAB and AMBER12 keywords. Specifies that the expected chirality of each centre should be
maintained based on that in the initial quenched minimum, rather than the standard chiralities found in most proteins/nucleotides.
WARNING: Presently for AMBER12, without this keyword being included, no chirality checking is done at all!
- SC nn mm sig sceps scc: specifies a Sutton-Chen potential[#!suttonc90!#] with
parameters n =nn, m =mm, a=sig, ε =sceps and
c =scc.
- SEED nsstop: if the SEED keyword appears then the program
looks for a file seed containing coordinates, which are used to `seed' the new run.
The number of coordinates given in this file should be no more than one less than the number
given in coords. The specified coordinates are frozen from the first step until
step nsstop.
- SHIFTCUT: specifies a shifted-truncated potential for bulk binary Lennard-Jones.
- SLOPPYCONV bgmax: specifies a basin-hopping run (as opposed to standard MC
on the untransformed surface). bgmax is the convergence criterion
for the RMS force in the basin-hopping
quenches. If this criterion is too strict then the run time will be greatly increased.
If it is too sloppy then the performance of the algorithm is impaired. Different values
are needed for different potentials. BASIN can be used instead.
- SORT: for pairwise potentials the atoms can be sorted from most to least
strongly bound. The SORT keyword enables this sorting for the coordinates printed
in file lowest. This can be useful for seeding subsequent runs by removing the
most weakly bound atoms. This sort is not set by default and is meaningless if the
pair energies are not computed.
- SPECLABELS L1 L2 ...LM : specifies the labels to be used in file lowest for each atomic species. Intended for use with a potential invoked by keywords MLJ, MGUPTA and MSC. Note that each label is interpreted as a string of two characters, and the calculation will stop if the supplied number of labels (M) does not match the species count.
- SPECMASS m1 m2 ...mM: specifies the mass for each atomic species (all masses are 1 by default). Intended for calculation of a mass-weighted Hessian or molecular dynamics simulations. Error will occur if the supplied number (M) of masses does not match the species count.
- SPRING type factor: specifies the use of a surrogate spring potential. For ReaxFF (or other potentials) we may need to restrain some bonds to a specified value to avoid reactions.
In such cases, the surrogate spring potential should be added between one or more pairs of atoms in the system. type specifies the type of spring: either harmonic or reaxff and
factor (default: 0.95) specifies a value that multiplies the spring constant(s) in each call to potential during SLOPPY quenches. The lowering of spring constant(s) can be used to
recover the original potential before the TIGHT (final) quench (the surrogate potential is switched off). A file called springfile is required to specify the pairs of atoms to be restrained
together with the associated spring constants and bond length restraints. The syntax of springfile is the following:
N
Ati Atj Kij Rij
Where N is the total number of listed bonds, Ati and Atj are atom ID's, Kij is a spring constant and Rij is a bond restraint length. In case of a reaxff type spring, an additional
spring constant should be listed immediately after the 1st spring constant. Values for the spring constants are potential and system dependent. For a ReaxFF system, a typical value for the 1st
(2nd) spring constant is 1E7 (2.5).
- STAR: specifies an excited state calculation for Ar*n or Ne*n for
a diatomics-in-molecules potential when used with NEON or ARGON.
- STEP step astep ostep block: specifies the maximum step sizes. step is
for the maximum change of any Cartesian coordinate and astep specifies a tolerance
on the binding energy of individual atoms (if available, i.e. for Morse and LJ) below
which an angular step is taken for that atom. See the following section for more details.
ostep is the maximum displacement of an axis-angle coordinate for a rigid body system
and block (an integer) is the block size for which separate translational and orientational
displacements will be made for rigid bodies. Omitting block or using a value of zero results in
translational and orientational steps being taken simultaneously
for rigid bodies. The default values for step,
astep and ostep are all 0.3 and the default value of nblock is zero.
If the system is in fractional coordinates, the step will automatically be adjusted to an appropriate length.
See also STEPRANGE for the allowed range when the maximum step is adjusted for a given acceptance ratio.
- STEPRANGE min max: specifies minimum and maximum values for the step size parameter
when steps can be adjusted to achieve a given acceptance ratio.
See also STEP and ACCEPTRATIO.
- STEPBOX length angle: specifies the maximum step sizes for cell parameter optimisation
in runs with BOXDERIV. length and angle are the maximum step sizes for cell lengths in the
prevailing units and angle in radians.
- STEEREDMIN smink sminkinc smindiststart smindistfinish sminatoma sminatomb: specified steered
minimisation should be performed (must be used with AMBER9). For a protein/ligand system, this adds a translation
to the MC move. The vector between the centre of coordinates of groups A and B (as defined in the file movableatoms)
is calculated and set to smindiststart. During the following minimisation, a restoring force is applied to
the ligand. The harmonic force constant is initially zero, and rises by sminkinc every LBFGS step up to a
maximum of smink. The force is applied until the A->B distance is less than smindistfinish.
- STEPS mcsteps tfac: determines the length of the
basin-hopping run through the integer mcsteps and the annealing protocol through
the real variable tfac. The temperature is multiplied by tfac
after every step in each run.
- STICKY nrbsites, sigma: specifies a `sticky patch' potential with nrbsites
sites in the rigid body reference and a value of sigma for the σ parameter.
- STOCK mu lambda: specifies a Stockmeyer potential with parameters
μ and λ, respectively.
- STOCKAA muD E: calls a finite system of Stockmayer particles,
where μD is the dipole moment strength and the electric field of stength E can be optionally present. The field, if
present, is along the space-fixed z-direction.
- STRAND: specifies a system of β strands coded using the rigid body formalism.
- STRESS mode: calculate and print atomic-level stresses in the file lowest. The integer parameter mode is optional, with the default value being 1, when the local pressure and the corresponding anisotropy parameter are printed after each atom's coordinates. Per-atom energy is also printed in the final field. Additional modes can be introduced if other invariants of the local stress tensor are desired.
- SUPPRESS: suppresses the majority of the GMIN output.
Think of it as the opposite of DEBUG.
- SW: specifies the Stillinger-Weber Si potential.
- SYMMETRISE int tol1 tol2 tol3 tol4 tol5 qmax mdiff d: specifies that the symmetrisation
routine should be called every int steps. The five tol parameters are tolerances
for various parts of the routine:
tol1 is used in ptgrp.f in defining orbits;
tol2 is the distance tolerance used in ptgrp.f to define point group symmetry operations;
tol3 is the maximum relative difference in principal moments of inertia used to
diagnose point groups with degenerate irreducible representations in ptgrp.f;
tol4 is the distance cutoff used to determine if a symmetry element has been lost in symmetry.f.
Since we are dealing with approximate symmetries, this parameter may be larger than tol2.
It is compared to the largest atomic displacement divided by the corresponding radius
for the closest permutation.
tol4 is also used to test whether atoms lie on a given symmetry element, and in testing
whether orbits generated from `floaters' are actually contained in the core.
tol5 is generally to check for atom clashes in symmetry.f, including analysis of
missing sites in orbits, as well as overlap between orbits generated from `floaters' and
previous core or new orbit sites.
qmax is the maximum number of quenches allowed for each call to symmetry.f.
mdiff is used to test whether a generated symmetry operation is new. If any of the nine
components of the corresponding 3×3 matrix differs by more than mdiff from an
existing matrix then the operations are considered to be different.
d is the exponential factor used in constructing a centre of mass that is biased towards
core atoms. The contribution of each atom is weighted by
exp(- dx(i)), where x is the
centre of mass distance of atom i on the previous cycle.
- TABOO nlist: specifies a taboo list of the nlist lowest minima should be maintained.
- TARGET target1 target2 ... : specifies any number of target energies.
The current run stops in an orderly
fashion if the current quench energy is within econv of any target or below (see EDIFF).
See also TARGETMATCH and FORBIDLIST.
- TARGETMATCH target1 target2 ... : specifies any number of target energies.
The current run stops in an orderly
fashion if the current quench energy is within econv of any target (see EDIFF).
This keyword can therefore be used to target specific local minima.
See also TARGET and FORBIDLIST.
- TEMPERATURE temp: defines the temperature, temp, at which the
MC runs are conducted. Different values can be specified for serial `parallel' runs if
PARALLEL is set.
For true parallel basin-hopping use the BHPT keyword and omit TEMPERATURE.
- TERMINALCAPPING n: bypasses cis-trans checks for the bonds formed between terminal groups (for example, ACE and NME) and standard amino acids. By default, these bonds are treated like normal peptide bonds, but this keyword ensures they are handled appropriately. n represents the number of capped monomers in a homo-oligomer (default value: 1).
- TETHER hdistconstraint hwindows ExtrapolationPercent lnHarmFreq: requests a calculation of the vibrational density of
states for a given minimum. hdistconstraint is the minimised average radius of the basin of attraction to which the minimum
belongs, hwindows is the number of potential energy windows into which a WL simulation is split. ExtrapolationPercent is the percentage of the whole potential energy spectrum, for which the density of states is estimated from
the harmonic approximation and not sampled. lnHarmFreq the log product of positive Hessian
eigenvalues.
- THOMSON q: specify the Thomson problem for unit charges on a sphere.
If q is present it is taken to be the charge on one particle, which can
therefore be different from all the other unit charges and is read as a real number.
- TIGHTCONV cgmax: cgmax is the tolerance for the
RMS force in the final set of quenches that are used to produce
the output for file lowest. The default is
cgmax= 10-3, but the appropriate values depend upon the system in question.
QMAX can be used instead.
- TIP n: specifies a TIPnP intermolecular potential for rigid body water molecules.
≤n≤5.
- TIP4PGR: specifies genrigid implementation of TIP4PGR intermolecular potential for rigid body water molecules.
Requires files coordsinirigid and rbodyconfig.
- TOLBRENT tolb: parameter for DBRENT minimisation.
Inefficient compared to LBFGS.
- TOMEGA: used with the CHARMM keyword to specify that peptide bonds will be twisted along with all other dihedrals.
- TOSI app amm apm rho: specifies the Tosi-Fumi potential[#!tosif64!#]
with parameters A++, A–, A+- and ρ.
- TRACKDATA: produces `energy.dat' and `markov.dat' containing the quench number and
associated energy and markov energy in two columns and `best.dat', containing the current quench number and the current lowest
total energy. If RMS is also specified, a file called `rmsd.dat' is produced containing the RMSD from a reference structure.
See RMS for more information. This allows plotting with gnuplot to monitor convergence of multiple runs.
If A9INTE is also specified, two additional output files are produced, `intE.dat' containing the quench number and associated interaction
enthalpy, and `bestintE.dat' containing the quench number and current lowest interaction enthalpy. This keyword does not yet function for MPI runs.
- TRANSLATERIGID freq maxdist: for use with the generalised rigid
body framework, RIGIDINIT. Randomly translates each rigid body in the
system by a distance up to maxdist every freq steps.
- TSALLIS q: specifies that steps are accepted/rejected using Tsallis statistics with the
given value of q, rather than the usual Boltzmann condition.
- TTM3: specifies Xantheas' TTM3-F water potential, coded
by Jeremy Richardson.
The atom order must be O1, O2,..., H1a, H1b, H2a, H2b,....
The energy is in kcal/mol and the distance unit is Å.
Hydrogens are NOT permutable between different oxygens.
Note that this potential is prone to cold fusion—setting the COLDFUSION
parameter explicitly may be necessary.
- TWIST n1 n2 n3 n4 f ref: adds a twist potential corresponding to the
torsional angle for atoms n1, n2, n3 and n4.
The functional form is
Vtwist(φ) = f sin(φ - φref.
phiref and the angular displacement are set to the range - π to π,
but with the sin function dependence this should now make no difference.
Setting
φref to the angle for the global minimum or native state might
be convenient.
- TWOPLUS: when combined with keywords NEON or ARGON
uses a diatomics-in-molecules potential for the doubly charged cation.
- UACHIRAL: MUST be included when using ff03ua, the AMBER united atom forcefield unless you have disabled the checks for inverted chiral carbons
with NOCHIRALCHECKS. UACHIRAL ensures the correct impropers are used to define sidechain chirality when HB hydrogen is missing.
- UNIFORMMOVE: the proposed steps for each atom in takestep are uniformly
distributed over a sphere whose radius is adjusted on-the-fly to give the required acceptance
ratio (or fixed if FIXSTEP or FIXBOTH is set).
The default behaviour for backwards compatibility is for the separate x, y and z displacements
of each atom to be scaled uniformly within the current step size range.
- UCC nops hilbert maxops: Unitary coupled cluster potential with nops active operators taken from the
first entries in a pool of maxops possibilities (default is that all nops operators are used).
hilbert is the dimension of the hilbert space.
This keyword requires additional files ham for the Hamiltonian,
dimension hilbert × hilbert,
and opmat, with dimensions nops × hilbert × hilbert.
See UCCGBH for optimsation of operator order using a generalised basin-hopping approach.
- UCCGBH interval rms maxlbfgs swaprange: generalised basin-hopping to optimise operator order using
swaps of operators to define changes within the current neighbourhood.
swaprange is the maximum number of operators to attempt swaps to from the pool, and must be
less than maxops.
Additional files of type oporder with the operator order corresponding to entries in final lowest
files will be printed. The inital order is read from oporder
See keyword UCC.
- UCCHIGHEREX: Works alongside QC1 and QCUCC, specifies triple and quadruple excitations.
Instead of the standard four-column format for qc_ucc.dat, use eight lines instead.
Not compatible with ANSATZ.
- UPDATES nup: UPDATES is the number of previous steps saved in the LBFGS routine,
default 4.
- UPDATERIGIDREF: Updates the rigid body reference coordinates after a step has been taken when using
RIGIDINIT. This allows steps to be taken within rigid bodies if required. As unphysical conformations may be introduced
into the rigid bodies this way, HYBRIDMIN should be allowed these to be relaxed.
- USEFRAC: For CASTEP and VASP the USEFRAC directiove specifies that the atomic coordinates are
manipulated in fractional form during the optimisation.
This option is on automatically if BOXDERIV is set.
- USEFRQS: use normal mode frequencies for CNBH if a quantum vibrational
partition function is required. Accesses file min.frqs. Without this keyword the high temperature
classical vibrational partition function is used in CNBH and FEBH.
- USEROT: Include the rigid rotor partition function during a free energy basin-hopping run.
- USEMINDATA n: create a min.data file for use with CNBH. If USEFRQS is set
then a min.frqs file (direct access) is also created. Unless NOPOINTS is set a points.min
file will also be curated.
The min.data file has the same format as for PATHSAMPLE but there is an
extra column specifying the number of atoms.
n is the maximum number of atoms. This value is needed to set the direct access record size for
min.frqs and points.min files.
The maximum size set for GCBH needs to be smaller than n.
- VGW ljsigma ljepsilon taumaxsg taumaxfg: Specifies use of VGW quantum quenching in place of
classical minimization routines such as LBFGS. ljsigma and ljepsilon are the corresponding Lennard-Jones
parameters that must be specified, and taumaxsg and taumaxfg are the maximum value of ``imaginary'' time τ (inverse tempertaure) for the propagation.
The former pertains to the faster ``single-particle'' SP-VGW used for quenching during the MC runs, and the latter for the more accurate
``fully-coupled'' VGW used for the final quenching (analogous to the tight convergence of the LBFGS). A τ of at least
2.5 is recommended for the SP-VGW and 5.0 for the FC-VGW. A file vgwdata containing the masses (in a.m.u.) of all particles, in order of the location
of their xyz coordinates in ``coords'' must be present (e.g. for a 38 atom Ne cluster, vgwdata will have 38 lines of ``20''). Different
masses are permitted, though the current version allows for only one set of LJ parameters.
- VGWCPS on magnitude: Specifies use of contraining potential for SP-VGW (sloppy convergence), as clusters expand during quantum quenching
with decreasing mass. 1 or 0 for on corresponds to on/off,
and magnitude should range from 1 to 1000, with 1 having minimal effect, 1000 being highly constrained. Default value is ``on'', with magnitude 1.
- VGWCPF on magnitude: Same as VGWCPS but for FC-VGW, used for the final, full quenching (tight convergence).
- VGWTOL magnitude: Absolute tolerance parameter for differential equation solver used for VGW quenching. Default value is 0.0001.
For highly quantum or ``stiff'' systems this may need to be increased, while it may be decreased for ``softer'' or less quantum systems to enhance
speed.
- VISITPROP: if specified the Wang-Landau convergence is governed by proportionality of visits to the current value of
the modification factor, and not the histogram flatness criterion [#!ZhouB03!#].
- WELCH
A++ A– A+- ρ Q+ Q- α+ α-: specifies a Welch binary
salt potential with the parameters indicated.
- XTB system runparams: Provides an interface to the xtb program as subprocess; see
the documentation at xtb-docs.readthedocs.io/en/latest/contents.html.
system is the file to read input coordinates from; runparams is a string
with options for the xtb executable, which must be in the current path. For example:
XTB water21 '–acc 0.1 –gfn2 -P 8'
xtb can fail when the SCC calculation does not converge. In this case GMIN sets the cold fusion
condition to escape from the current quench.
Note: GMIN assumes that the coordinates are in Å, but xtb retruns the gradient in hartree/ bohr, so there is a conversion factor in the gradient. system can be in either coord (turbomole), .xyz or .gen format, the interface will automatically account for the unit conversion to Å. If system is a start file, additionally a file xtb.symbols is required containing the element symbols (one per line).
More keywords associated exclusively with the XTB keyword:
Note: if you use gfnff then xtb uses the coordinates to define a tolopogy and the hence the gfnff potential.
If the geometry changes enough, the topology and the potential can change, making the energy discontinuous
and not reproducible on restart. The solution to this problem is to use XTBAPI instead.
- XTBCONTROL file: Allows xtb to read in additional instructions from a separate file, as for example GBSA/ALPB grid sizes.
- XTBPATH bin: Allows to specify another xtb binary bin. By default 'xtb' is used.
- XTBAPI system level charge uhf: Provides an interface to the xTB
methods. Requires to build the XTBGMIN binary to be build with -DWITH_XTBAPI=true. The respective git submodules must be loaded for
this. system is the file to read input coordinates from; level is
either of { gfn1, gfn2, gfn0, gfnff }. charge is an integer
specifying the molecular charge. uhf is an optional parameter used to
specify the number of
nα - nβ electrons, which for closed shell systems is zero and can be omitted.
The setup has been changed to allow a consistent reference geometry to be read from the system file,
so that gfnff calculations can be run with a potential that does not change with geometry. After initialisation,
the coordinates in system will the overwritten from coords or from a dump file if
RESTORE is requested.
An additional keyword associated with XTBAPI is:
- XTBARG argument value: Modifies the setting argument and sets it to value for the xtb calculator. Available arguments are:
| argument |
value |
description |
| gbsa or alpb |
〈solvent name〉 |
Instructs the setup of GBSA or ALPB implicit solvation. For available solvents see the xtb documentation |
| accuracy |
〈real number〉 |
Increase/decrease SCC accuracy. Default is 1, values smaller than 1 increase the accuracy |
| scc |
〈integer〉 |
Set the maximum number of SCC cycles |
| etemp |
〈real number〉 |
Set the electronic temperature (in K) for Fermi smearing of orbital populations |
To build XTBGMIN with Intel
load only the appropirate modules:
cmake/3.23.2 tbb/64/2022/0/1 icc/64/2022/0/1 gcc/12.2.0 idb/latest oneapi-compiler-rt/64/2022/0/1 mkl/64/2022/0/0
Always specify the compiler environment variables when configuring the build
FC=ifort CC=icc CXX=icc cmake ~/softwarewales/GMIN/source/ -DWITH_XTBAPI=yes
- ZETT1 and ZETT2: specify the Zetterling potentials.
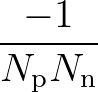
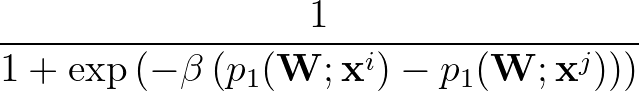 .
.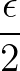

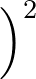 +
+ 
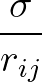
![$\displaystyle \left.\vphantom{ \left(\frac{r_{ij}}{\sigma}\right)^2+
\left(\frac{\sigma}{r_{ij}}\right)^2 }\right]$](img34.png) ,
,
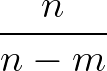
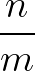
 ε
ε
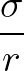
 -
- 
![$\displaystyle \left.\vphantom{ \left(\frac{\sigma}{r}\right)^{n} - \left(\frac{\sigma}{r}\right)^{m}}\right]$](img55.png)
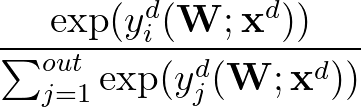
![\includegraphics[width=0.5\textwidth]{PHE.eps}](img70.png)
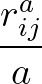 -
- 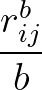 ,
, 1 +
1 + 
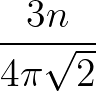
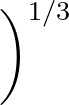
![$\displaystyle \left.\vphantom{1 + \left(\frac{3 n}{4\pi\sqrt{2}}\right)^{1/3}}\right]$](img77.png) ,
,