

Next: Specification of Potentials
Up: OPTIM2 User Guide
Previous: Changes from OPTIM
Keywords
The following keywords are recognised, where n and x are integer and
real data, respectively.
- 2D: restrict the system to the
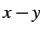 plane.
plane.
- ADM n: will cause the
interatomic distance matrix to be printed every n cycles;
the default for n is 20. This matrix is not printed by default.
- AMBER: specifies a calculation with the AMBER potential. This requires
auxiliary files amber.dat and coords.amber in the same directory.
- AXIS n: specifies the highest symmetry axis to search for in
routine symmetry; default is six.
- AXTELL
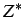 : specifies that the axilrod-Teller potential with
coefficient is to be added to the potential specified by the atom type.
: specifies that the axilrod-Teller potential with
coefficient is to be added to the potential specified by the atom type.
- BFGSCONV gmax:
gmax is the convergence criterion
for the root-mean-square gradient, default
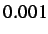 .
This is also the convergence criterion
for the subspace minimisations in hybrid EF/BFGS transition state searches, for use with BFGSTS.
.
This is also the convergence criterion
for the subspace minimisations in hybrid EF/BFGS transition state searches, for use with BFGSTS.
- BFGSMIN gmax: instructs the program to perform a BFGS minimisation.
gmax is the convergence criterion
for the root-mean-square gradient, default .
- BFGSSTEP: instructs the program to step off a saddle point along the
eigenvector corresponding to the smallest negative eigenvalue without
diagonalizing the Hessian to find this eigenvector. It is possible to step off
parallel and antiparallel to this eigenvector by specifying positive or negative values
for the MODE parameter. BFGSTS need not be set, but see also
PATH and CONNECT.
- BFGSSTEPS n: sets the maximum number of steps allowed in an LBFGS minimisation.
Default value is the value of the STEPS parameter.
- BFGSTS nevs ncgmax1 ncgmax2 ceig nevl: instructs the program to perform a
hybrid BFGS/eigenvector-following transition state search.
nevs is an integer that defines the largest number of iterations allowed in the
calculation of the smallest Hessian eigenvalue; default 500.
ncgmax1 is an integer that defines the largest number of BFGS steps
allowed in the subspace minimisation before the eigenvalue is deemed to have converged; default 10.
ncgmax2 is an integer that defines the largest number of BFGS steps
allowed in the subspace minimisation after the eigenvalue is deemed to have converged; default 100.
The eigenvalue is deemed to be converged if the modulus of the overlap between the corresponding
eigenvector and that saved from the previous step exceeds 0.999 and we have the right number of
negative eigenvalues.
ceig is a double precision parameter that sets the convergence criterion for
the calculation of the smallest non-zero Hessian eigenvalue.
If NOHESS is set ceig is compared to the RMS `force' corresponding to the
Rayleigh-Ritz
expectation value that is minimised to get the smallest eigenvalue.
If the Hessian is used via iteration then ceig is compared to the percentage change in the eigenvalue between successive
steps. The default value is 1.0, which is more appropriate to the latter case. Smaller
values are probably necessary in BFGS/BFGS calculations.
nevl is an integer that defines the largest number of iterations allowed in the
calculation of the largest Hessian eigenvalue; default 100. Not needed if NOHESS
is set.
- BINARY ntypea epsab epsbb sigmaab sigmabb: specifies a binary Lennard-Jones
system for use with the Lp or Ls atom types. ntypea is the number of type
A atoms--the rest are assumed to be type B and appear at the end of the list
of coordinates.
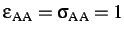 define the units of energy and length,
and epsab=
define the units of energy and length,
and epsab=
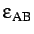 , epsbb=
, epsbb=
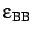 ,
sigmaab=
,
sigmaab=
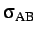 , sigmabb=
, sigmabb=
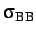 .
.
- BSMIN gmax eps: calculates a steepest-descent path using gradient only
information with convergence criterion gmax for the RMS force and initial
precision eps. The Bulirsch-Stoer algorithm is used.
- CADPAC system exec: tells the program to read derivative information in
CADPAC format. system is a string to identify the system and exec is
the name of an executable that will generate a CADPAC input deck from a points file.
If exec is omitted its name is assumed to be editit.system.
- CALCRATES temp h: rate constants will be calculated for each transition state
found in a CONNECT or PATH run, or using saved information in an
old path.info file if READPATH is specified. Both forward and backward canonical
rate constants are calculated from transition state theory at temperature temp (default
temp=1), and values are given
for both quantum and classical harmonic vibrational partition functions. h is the
value of Plank's constant in suitable reduced units for the corresponding potential (default value 1).
- CASTEP system: tells the program to use CASTEP to calculate energies and
forces with input files based on the system name system.
- CHECKINDEX nevs ceig nevl: instructs the program to check the Hessian
index after a BFGS minimisation or BFGS hybrid transition
state search. This keyword also works with NOHESS.
The parameters are the same as for BFGSTS below, and need not be set
if they have already been specified by that keyword.
- CHECKCONT: if CHECKINDEX is specified then CHECKCONT
instructs the program to take a pushoff and continue if convergence to a stationary
point with the wrong Hessian index is detected.
- COMMENT or NOTE: the rest of the line is ignored.
- CONNECT n: find a min-sad-min-
 -min sequence connecting
the initial minimum in odata to the final minimum in finish,
giving up if more than n transition states are needed (default n=100). CONNECT
generally needs to be augmented by other keywords to specify how the transition
state geometries should be guessed (NEB or FIXD), how the transition
state searches should be performed (SEARCH 2 or BFGSTS, with or
without NOHESS, NOIT etc.), and how that pathways should be
calculated (SEARCH 0, 6 or 7, BFGSMIN, RKMIN or BSMIN).
A summary file will be produced if DUMPPATH is specified. An xyz file
for the overall path will be printed to path.xyz and the energy as a
function of the path length is printed to EofS. The corresponding xyz and energy
files for the individual steps in the path are numbered path.
-min sequence connecting
the initial minimum in odata to the final minimum in finish,
giving up if more than n transition states are needed (default n=100). CONNECT
generally needs to be augmented by other keywords to specify how the transition
state geometries should be guessed (NEB or FIXD), how the transition
state searches should be performed (SEARCH 2 or BFGSTS, with or
without NOHESS, NOIT etc.), and how that pathways should be
calculated (SEARCH 0, 6 or 7, BFGSMIN, RKMIN or BSMIN).
A summary file will be produced if DUMPPATH is specified. An xyz file
for the overall path will be printed to path.xyz and the energy as a
function of the path length is printed to EofS. The corresponding xyz and energy
files for the individual steps in the path are numbered path.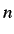 .xyz and EofS. for
transition state .
.xyz and EofS. for
transition state .
- CONVERGE x y: overrides the default convergence conditions
for eigenvector-following and second order steepest-descent calculations
described in §6. If one number is supplied then convergence depends only upon
the maximum unscaled step falling below the specified value (and the right number
of negative Hessian eigenvalues). If two numbers are supplied the second is the
required threshold for the RMS force which must also be satisfied.
- CPMD system: tells the program to use CPMD to calculate energies and
forces with input file system; bulk boundary conditions.
- CPMDC system: tells the program to use CPMD to calculate energies and
forces with input file system; cluster boundary conditions.
- CUBIC: if the three box lengths are equal to start with in a PV run,
and the keyword CUBIC is included, then a cubic box is maintained.
- DCHECK ON/OFF: turns on/off warning messages when atoms
approach to within
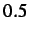 distance units units. Default is ON.
distance units units. Default is ON.
- DEBUG: causes unscaled steps, gradients, Hessian eigenvalues and
summary information to be printed every cycle for SEARCH 0-7 runs. Turned off by default.
Also produces much more printing for individual steps in CONNECT runs.
- DFTB: specifies that the tight-binding code of Walsh and Wales[11]
is used.
- DGUESS dguess1 dguess2: initial guess for diagonal elements of the inverse
Hessian, used whenever the BFGS optimiser is reset. dguess1 is used in
geometry optimisations, dguess2 is used for minimisation in eigenvalue
calculations.
When a Hessian is
available (i.e. for an EF/BFGS calculation) dguess1 is estimated by the
program. Otherwise the default values are dguess1=dguess2=0.1.
- DOUBLE: adds a double-well potential as per J. Chem. Phys.,
110, 6617, 1999. The
energy and derivatives add to the potential specified by the atom
type. Only atom type
`LS' (not binary) has the 1,2 interaction removed as well.
At low density some atoms may not interact with any others and
problems result due to additional zero eigenvalues.
- DUMPPATH: prints a summary of the min-sad-min--min path produced by CONNECT to
file path.info. For each stationary point the energy, point group order and symbol,
Hessian eigenvalues and coordinates are given. Hessian eigenvalues are computed
if not yet calculated, otherwise they are saved during the CONNECT process.
- DUMPVECTOR [ALLSTEPS] [ALLVECTORS]: if present the Hessian eigenvectors
will be written to file vector.dump after each OPTIMisation step,
preceded by the corresponding eigenvalue for each one. The
eigenvectors corresponding to zero eigenvalues are excluded. For hybrid eigenvector-following/BFGS
transition state searches only the results corresponding to the smallest non-zero
eigenvalue are dumped. The default is to dump only the vector corresponding
to the smallest non-zero eigenvalue for the last step. If ALLSTEPS is
present then vectors are dumped at every step. If ALLVECTORS is present
then all the vectors corresponding to non-zero eigenvalues are dumped, rather
than just one of them. ALLSTEPS and ALLVECTORS can be present in
either order.
- EFSTEPS n: prints the unscaled steps in the Hessian
eigenvector basis every n steps--turned off by default.
- EVCUT x: sets a cutoff value for eigenvalues. Eigenvalues
of magnitude less than
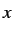 are treated as zeros and steps along such directions
are omitted.
are treated as zeros and steps along such directions
are omitted.
- FAKEWATER: specifies that implicit water should be used in an AMBER
calculation via an effective dielectric constant.
- FILTH n: specifies an integer variable, n, to distinguish output files in
parallel landscape jobs.
- FIXD t12fac dthresh: transition state search using hard sphere moves. A random
velocity vector is assigned and the first hard sphere collision is located. If t12fac
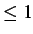 then the system is moved through a fraction t12fac of this first collision time; the
default value is 1. If
t12fac
then the system is moved through a fraction t12fac of this first collision time; the
default value is 1. If
t12fac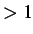 then the two atoms are moved half way between the entrance and exit of their
collision, but their separation is rescaled to unity. See also RANSEED.
Used in CONNECT procedure only if FIXD is set and the two
minima to be connected are closer than dthresh.
then the two atoms are moved half way between the entrance and exit of their
collision, but their separation is rescaled to unity. See also RANSEED.
Used in CONNECT procedure only if FIXD is set and the two
minima to be connected are closer than dthresh.
- FIXAFTER n: specifies that FIXIMAGE should be set permanently after step
n. This effectively freezes the interacting images in different supercells
for calculations with periodic boundary conditions.
- FREEZE n1 n2 etc.: specifies that atoms n1, n2, etc. should be frozen.
Only works for LBFGS minimisation and BFGSTS if 3 or more non-linear atoms are frozen.
- FRACTIONAL: specifies fractional coordinates to be used in box length
optimisation for constant pressure runs using PV.
- GAMESSUS: potential will be generated by GAMESS-US.
- GAMESSUK: potential will be generated by GAMESS-UK.
- GAUSSIAN: tells the program to read derivative information in
Gaussian92 format.
- GDIIS x y z: initiates geometry direct inversion of the
invariant subspace. This option always seems to give poorer convergence
if analytic second derivatives are available at every step. It requires
three real parameters, which are read after the GDIIS keyword. The
first is the RMS force below which GDIIS will be used, the second is the
dimension of the DIIS problem to solve and the third is the interval between
GDIIS steps.
- GRADIENTS n: causes the gradients along the Hessian
eigenvectors to be printed every n cycles. Gradient printing is turned off by default.
- GRADSQ gsthresh nspecial nallow specifies optimisation of the modulus gradient
with nallow second order steps at intervals of nspecial if the RMS force lies below gsthresh.
- HUPDATE nstart ninterval phi: uses a Hessian update
procedure instead
of calculating the analytic second derivatives at every step.
nstart is the first step at which analytic derivatives will be calculated; by
default nstart=0, the Hessian is initialised to the identity and no
analytic second derivatives are ever calculated. ninterval is the interval
at which analytic Hessians are calculated; the default is 0, which means that no
additional analytic Hessians are ever found. A transition state search starting from
a minimum seems unlikely to work unless we calculate analytic Hessians at regular
intervals including the first step. Update applied is
phi time Powell[12] plus (1-phi) times Murtagh-Sargent.
(See Bofill and Comajuan, J. Comp. Chem., 11, 1326, 1995.)
- INDEX n: initiates a search for a saddle of index n if
SEARCH 2 is specified. See also KEEPINDEX. Also works with BFGSTS
up to a maximum of index 50, but NOIT must be set and a Hessian is needed.
- KEEPINDEX : specifies that INDEX is set equal to
the number of negative Hessian eigenvalues at the initial point.
- MASS: Specifies the use of mass-weighting for the steps; the input
and output coordinates are not affected.
- MAXBFGS x1 x2 x3 x4: x specifies the maximum allowed step length in LBFGS
minimisations, x1 for normal minimisations, x2 for eigenvector optimisation
by minimisation, x3 for putting structures in closest coincidence with
mind, and x4 for NEB minimisations. Default values all 0.2.
- MAXSTEP x1 x2: specifies the maximum step size for eigenvector-following and
steepest-descent calculations, default value 0.2. The precise
implementation of this constraint depends upon the step scaling method employed (see §9).
- MAXMAX x: specifies the maximum value that the maximum step size
is allowed to rise to. The default value is .
- MINMAX x: specifies the minimum value that the maximum step size
is allowed to fall to. The default value is
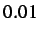 .
.
- MODE n1 n2: in an eigenvector-following transition state search specifies the eigenvector
to be followed uphill, where 1 means the softest mode, 2 means the next softest, and so on.
Setting n1 to 0 means that the softest mode is followed uphill at every step,
otherwise a maximum overlap criterion is used to determine the mode followed after the
first step. The sign of n1 is used to determine the direction in which we
step along the eigenvector if we are starting from a stationary point. This enables one
to start transition state searches from minima along any of the eigendirections in
either sense. If a minimisation is started from a converged transition state then setting
n1 to
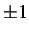 enables one to walk down the two sides of the pathway. This is true for
runs based on BFGS techniques too if the keyword BFGSSTEP is used.
MODE is now also used in EF/BFGS searches so long as an analytical Hessian is
calculated. n1 is used for the first step and n2 for subsequent steps. The
default is 0 for both parameters. Note that n2 must be explicitly set equal to
n1 to follow the same mode (as judged by overlap) in following steps.
enables one to walk down the two sides of the pathway. This is true for
runs based on BFGS techniques too if the keyword BFGSSTEP is used.
MODE is now also used in EF/BFGS searches so long as an analytical Hessian is
calculated. n1 is used for the first step and n2 for subsequent steps. The
default is 0 for both parameters. Note that n2 must be explicitly set equal to
n1 to follow the same mode (as judged by overlap) in following steps.
- MOVIE: a movie will be dumped in xyz format for the AMBER potential.
- NEB nsteps nimage rmsneb: specifies a nudged elastic band pathway
calculation[13,14] using a maximum of nsteps steps with
nimage images and RMS convergence criterion rmsneb. The default values are
nsteps=100, nimage=10, rmsneb=0.1.
- NOIT: for use with hybrid eigenvector-following runs when a Hessian is available.
Specifies that the required lowest eigenvalue and eigenvector be found using
LAPACK routine DSYEVR instead of by iteration.
- NOHESS: perform a hybrid EF/BFGS transition state search without a
Hessian. The smallest eigenvalue and the corresponding eigenvector are found from a separate
minimisation problem for every combined EF/BFGS step.
- NORESET: specifies that periodic images should not be returned to the
primary supercell for calculations involving periodic boundary conditions. (May be
useful for calculating transport properties with kinetic Monte Carlo).
- NOPOINTS: printing of coordinates to file points for intermediate steps
is turned off.
- PARALLEL nproc: specifies that auxiliary programs such as GAMESS, CASTEP and
CPMD use nproc processors.
- PARAMS: enables additional parameters to be set for
specific potentials, e.g. for LJAT,
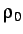 for Morse, n, m, the
box lengths
for Morse, n, m, the
box lengths 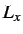 ,
, 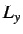 and
and 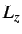 and the cutoff for ME, the
box lengths , and and the cutoff for JM, SC and P6,
and
and the cutoff for ME, the
box lengths , and and the cutoff for JM, SC and P6,
and 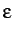 ,
, 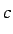 and
and 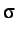 for Au, Ag and Ni. Note that all the
numbers are read as real.
for Au, Ag and Ni. Note that all the
numbers are read as real.
- PATH n: requests calculation of the pathway connecting two minima from the transition
state specified in odata. n is the number of intermediate points files to save on either
side, default n=3. A complete xyz file is printed to path.xyz and the energy as a function of
path length is printed to file EofS. A summary of the path characteristics such as barrier heights,
path lengths and cooperativity indices is printed at the end. If READVEC is present in
odata then the required eigenvector corresponding to the displacement direction will
be read from vector.dump, otherwise it will be calculated by diagonalizing a Hessian (for
SEARCH 2) or using an iterative approach (for BFGSTS with or without
NOHESS; NOIT can also be used with BFGSTS.)
After the initial step the energy will be minimised by whichever first or second
order method is specified in odata, i.e. eigenvector-following or second-order Page-McIver
for SEARCH 0 and 6 and 7, LBFGS for BFGSMIN, and first order steepest-descent
for BSMIN and RKMIN. See also DUMPPATH.
- POINTS: this must be the last keyword and introduces the
coordinate information which is read one atom per line. Each line must consist
of the atomic symbol of the atom followed by its three Cartesian coordinates.
Leading spaces before the atomic symbol should be avoided.
- PRESSURE: if present
tells the program to perform a constant zero pressure optimisation
for bulk SC, ME, MP, MS and P6. Normally a constant volume optimisation takes place.
- PRINT n: sets the print level. The default value is zero. Use
of this option is not recommended, since more specific print control is possible (see
below).
- PUSHCUT x: sets the threshold for the RMS force below which the
system may apply a PUSHOFF to escape from a stationary point of the wrong Hessian
index. Default value is 0.00001. Using a negative value prevents pushoffs from
occurring.
- PUSHOFF x: sets the magnitude of a step away from a stationary
point of the wrong order (see §9 for default action). Default value is 0.01.
Also used as the step size in transition state searches if the eigenvalue of the
direction being followed is positive.
- PV press gtol grad steps: specifies an approximate constant pressure optimisation with
pressure press and convergence criterion gtol for the gradients with
respect to the box lengths. If any box length gradient is larger in magnitude than
grad then up to a maximum of steps optimisation steps for the box lengths
are performed.
If the three box lengths are equal to start with and the keyword CUBIC is included,
then a cubic box is maintained.
Will not work with atom types MP, ME and MS.
Default values, press=0, gtol=0.001, grad
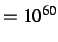 , steps=100.
, steps=100.
- PVTS press gtol grad nboxts: as for PV but searches for a transition
state with respect to box length nboxts, where nboxts=1, 2 and 3 specify the
,
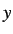 and
and 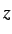 box lengths, respectively. nboxts defaults to 1.
box lengths, respectively. nboxts defaults to 1.
- RADIUS x: specifies that atoms should not be allowed to move beyond a distance x from the
origin.
- RANSEED n: specifies the initial seed for random number generation, as used in
hard sphere moves with FIXD.
- READPATH: if specified with CALCRATES then rate constants will be
calculated for the transition states found in path.info and no stationary point
searches will be performed.
- READVEC: if present the Hessian eigenvector
corresponding to the smallest eigenvalue
will be read from file vector.dump. The latter file can be generated using the
DUMPVECTOR keyword in a previous run.
- READHESS: Hessian will be read from file derivs at first step.
- REOPT: specifies that the eigenvector to be followed should be reoptimised
in a BFGSTS search after the EF step and before the tangent space minimisation.
- RESIZE x: scales all the coordinates by x
before the first step is taken. Default is 1.0.
- ROT: if present a rotational kinetic energy term is
added to the Hamiltonian. Either constant angular momentum (ROT JZ x) or
constant angular velocity (ROT OMEGA x) may be used to specify
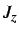 or
or 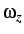 .
.
- RKMIN gmax eps: calculates a steepest-descent path using gradient only
information with convergence criterion gmax for the RMS force and initial
precision eps. A fifth order Runga-Kutta algorithm is used.
- SCALE n: sets the scaling algorithm as described in §9. The
default is n=10.
- SEARCH n: specifies the search type for eigenvector-following and
steepest-descent calculations, default is type 0.
The most common options are 0, a minimisation, and 2, a transition state search. See
§6 for full details.
- SHIFT x: specifies the shift applied to eigenvectors corresponding
to normal modes that conserve the energy. The default is
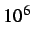 . The shift must be
large enough to move the eigenvalues in question to the top of the spectrum.
. The shift must be
large enough to move the eigenvalues in question to the top of the spectrum.
- STEPMIN n: sets the minimum number of optimisation steps before convergence
will be tested.
- STEPS n: sets the maximum number of optimisation steps per call
to OPTIM. The default is n=1. Setting n to zero at the start of a run
means that only a point group analysis is performed.
- STOPFIRST: in a CONNECT run, stop as soon as the initial minimum has a transition state
connection.
- SUMMARY n: prints a summary of the steps and derivatives
every n steps. The default is
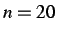 .
.
- SYMCUT x: specifies the RMS force below which the point
group symmetry subroutine will be called. The default is x=0.001.
- TOLD x: sets the initial distance tolerance for points
to be considered the same after rotations and reflections in point
group determination. The default is 0.0001.
- TOLE x: sets the initial tolerance for eigenvalues
of the inertia tensor to be considered equivalent in point
group determination. The default is 0.0001.
- TOSI
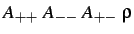 : specifies a Born-Mayer potential
with the parameters indicated (see §5).
: specifies a Born-Mayer potential
with the parameters indicated (see §5).
- TRAD x: sets the trust radius (see §9). The default value is 2.0.
- TWOD : specifies an optimisation in two dimensional space.
- UPDATES mupdate1 mupdate2: specifies the number of updates in the LBFGS routine before
resetting. mupdate1 is used for geometry optimisations and mupdate2 is used
in eigenvalue calculations. The default value is 4 for both parameters.
- VALUES n: prints the Hessian eigenvalues every n steps.
The default is n=20.
- VARIABLES: optimises a general function (which must be specified in
potential. The initial values of the variables are read one per line after the POINTS
keyword in odata.
- VECTORS n: prints the Hessian eigenvectors every n
steps. Eigenvector printing is turned off by default.
- WELCH
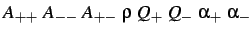 : specifies a Welch binary
salt potential with the parameters indicated (see §5).
: specifies a Welch binary
salt potential with the parameters indicated (see §5).
- ZEROS n: sets the number of zero Hessian eigenvalues to be
assumed--useful for general optimisation problems specified by VARIABLES.
Next: Specification of Potentials
Up: OPTIM2 User Guide
Previous: Changes from OPTIM
David Wales
2002-10-28