Input is keyword driven with sensible defaults in most cases.
Free format may be used within each line. Blank lines are ignored.
- 2D: enforce two-dimensional `flatland'.
- A9INTE: specifies that after each quench that does not lead to an inversion of chirality,
isomerisation of a peptide bond or cold fusion - the interaction enthalpy between a specified residue and the rest of the system
should be calculated using the external script `AMBGMINintE.sh', and read back into GMIN. This is intended for
use with protein/ligand systems where you are searching for low energy docked structures. As the total energy
does not fully correlate with the protein/ligand interaction enthalpy, it is often useful to retain not only the
lowest SAVE total energy structures, but also the lowest SAVEINTE interaction enthalpy structures.
To use this keyword, the `AMBGMINintE.sh' script (contained in the SVN repository in the SCRIPTS directory) must
be present in the GMIN working directory. You should ensure that you have edited it to match the residue numbering
of your system. You also need a full AMBER9+ installation with access to the `sander' executable. When using this
keyword, an interaction enthalpy dump file is produced every DUMPINT steps, and at the end of the run,
structural output files are produced for the SAVEINTE lowest interaction enthalpy geometries. After each quench,
the structure with the current lowest interaction energy is dumped in pdb and rst format prefixed with `bestint.' to allow
monitoring.
- ACCEPTRATIO accrat: accrat is the required acceptance ratio for the MC
exploration of the transformed surface. For fixed temperature runs (the default) the maximum step size
is adjusted to try and meet the requested value of accrat; for a fixed maximum
step size the temperature is adjusted instead. The default value of accrat is a half.
- Ackland id: specifies an Ackland embedded atom metal potential
coded by Dr Mihai-Cosmin Marinica.
id specifies the particular metal: 1 is ?, 2 is ?, 3 is ?, 4 is ?, 5 is iron, 6 is a different iron,
7 is tunsten.
Positive values for id specify periodic boundary conditions, where box lengths must be
specified by the PERIODIC keyword.
Negative values for id specify a cluster calculation. A CUTOFF value can also
be used for clusters.
- ALGLUE: specifies a glue potential for aluminium.
- AMBER9 inpcrd inpcrdformat: specifies a calculation with the interfaced
version of the Amber 9 program package. From this package the Amber force fields
are being used, with small modifications (e.g. smooth cut-offs).
Starting coordinates do not need to be specified in the odata file, they
are read from inpcrd instead (default coords.inpcrd), in Amber inpcrd
file format specified by the second optional argument inpcrdformat.
If the second argument is missing, it is assumed that inpcrd contains
only three columns with the xyz coordinates of all atoms, in the same order
as in the topology file. To start a run with this interface,
several auxiliary files are required in the same directory: input coordinate file
coords.inpcrd, parameter topology file coords.prmtop,
input file to Amber containing force field specifications min.in, and, if
desired, a coordinate file different from coords.inpcrd containing
starting coordinates.
To turn on smooth cutoffs for the Generalised Born force fields, the keyword
ifswitch=1 has to be used in the &cntrl namelist block of min.in.
When using the AMBER9 keyword, any calculated second derivatives will be
numerical. If one wants analytical second derivatives, the NAB keyword
should be used instead, with the same syntax.
Additional keywords for the AMBER 9 runs are DUMPSTRUCTURES, AMBERMDSTEPS,
LIGMOVE, BLOCKMOVE and MOVABLEATOMS.
- AMCHNMAX: The maximum number of angles that will be changed by up to STEP during an
AMBER dihedral step. If this is not set or is set to zero, cartesian steps of maximum size STEP are taken
instead.
- AMCHNMIN: The minimum number of angles that will be changed during an AMBER dihedral step.
- AMCHPMAX: The maximum probability for a single angle to be twisted in an AMBER dihedral step.
- AMCHPMIN: The minimum probability for a single angle to be twisted in an AMBER dihedral step.
- ANGSTROM: specifies coordinates in Ångstrom for the FRAUSI
potential.
- ARGON: introduces a diatomics-in-molecules calculation for
a neutral, cationic or electronically excited argon cluster. See also
GROUND, PLUS, TWOPLUS and STAR.
- ARM arma armb: use the acceptance-ratio method (Bouzida et al., Phys. Rev. A,
45, 8894, 1992) to adjust the step size to achieve the requested
acceptance ratio. A scaling factor is calculated and applied to step, rotmax,
and/or transmax. The scaling factor is calculated according to
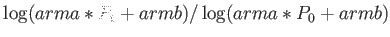 , where
, where 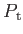 defines the
target acceptance ratio and
defines the
target acceptance ratio and 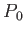 the actual acceptance ratio. Both values arma and
armb default to 0.4.
the actual acceptance ratio. Both values arma and
armb default to 0.4.
- ARNO: specifies a diatomics-in-molecules potential for Ar
 -NO clusters.
-NO clusters.
- AVOID dist maxsave: specifies that the geometry should be reseeded if the
latest structure gets within a distance dist of the maxsave members of a
cyclic list. If a third argument `F' is added to the AVOID line then such
steps are simply rejected rather than reseeded.
The NEWRESTART keyword must be specified to populate the list of
minima to be avoided. If the `F' argument appears for AVOID then
NEWRESTART will not reseed.
- AXTELL zstar: specifies an additive Axilrod-Teller term for certain
diatomics-in-molecules potentials as well as the Pacheco-Ramelho intermolecular potential for
C
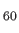 .[8]
zstar is the coefficient multiplying this term.
.[8]
zstar is the coefficient multiplying this term.
- BASIN bgmax: specifies a basin-hopping run (as opposed to standard MC
on the untransformed surface). bgmax is the convergence threshold
on the RMS force in the basin-hopping
quenches. If this criterion is too strict then the run time will be greatly increased.
If it is too sloppy then the performance of the algorithm is impaired. Different values
are needed for different potentials. SLOPPYCONV can be used instead.
- BASWAP Nwait Frac Nswaps: After a waiting period of Nwait steps, the coordinates of up to Nswaps unlike atom pairs (picked completely at random) will be exchanged with probability Frac. (This will happen instead of the random cartesian displacement move.) If Nswaps = 0, then just a single pair of unlike atoms will exchange coordinates, and in the selection procedure each atom is weighted by a Boltzmann factor with the corresponding potential energy change as the argument. This scheme requires the knowledge of all per-atom energies in the current state, as well as the energy each atom will have if it (alone!) changes type. [NOTE: This is tailored for binary atomic systems interacting via a pair-potential, such as BLJCLUSTER_NOCUT and BGUPTAT, but can also be applied to any other binary system with 0 < NTYPEA < NATOMS.]
- BASWAPTEST: prints useful information for benchmarking the effect of atom identity swaps in binary systems.
- BFGS: specifies that the full BFGS minimiser should be used. Inefficient compared to LBFGS.
- BGUPTAT NTYPEA AAA PAA QAA ZAA R0AA: One of the required keywords to specify a Binary Gupta run.
NTYPEA is specified, followed by the potential parameters for the A-A interactions. See also BGUPTATAB and BGUPTATBB.
[NB: Per-atom energies will be stored.]
- BGUPTATAB AAB PAB QAB ZAB R0AB: The line to specify the potential parameters for the A-B interactions
for a Binary Gupta run.
- BGUPTATBB ABB PBB QBB ZBB R0BB: Specifies the B-B interaction parameters.
- BHPT pttmin pttmax exchprob (random
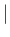 interval) (singlesets):
specifies a parallel tempering basin-hopping run with temperatures exponentially distributed between pttmin and
pttmax. Either the probability of attempting replica exchange (float,
interval) (singlesets):
specifies a parallel tempering basin-hopping run with temperatures exponentially distributed between pttmin and
pttmax. Either the probability of attempting replica exchange (float,
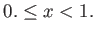 ) or the corresponding mean frequency
(integer,
) or the corresponding mean frequency
(integer, 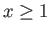 ) may be supplied as exchprob. Exchanges may be attempted at random or at intervals (default: random).
At each exchange event, either a single exchange between a random pair adjacent replicas is attempted, or exchange between all
pairs in either the set of even pairs,
) may be supplied as exchprob. Exchanges may be attempted at random or at intervals (default: random).
At each exchange event, either a single exchange between a random pair adjacent replicas is attempted, or exchange between all
pairs in either the set of even pairs,
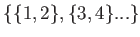 or set of odd pairs,
or set of odd pairs,
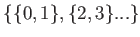 (default: single).
Should be used together with the MPI keyword.
(Only available if the source is compiled with MPI enabled.) If DUMPSTRUCTURES is specified, then every DUMPINT steps, minima from each replica will the written out in XYZ format to files dumpmin.replicaNumber.minimaNumber.
(default: single).
Should be used together with the MPI keyword.
(Only available if the source is compiled with MPI enabled.) If DUMPSTRUCTURES is specified, then every DUMPINT steps, minima from each replica will the written out in XYZ format to files dumpmin.replicaNumber.minimaNumber.
- BLOCKMOVE nblocks block_1 block_2 ... block_n: used with AMBER9, MOVABLEATOMS and LIGMOVE.
Divides the atom list in the ‘movableatoms' file into nblocks distinct blocks that are treated independently as
rigid during steptaking moves. block_i specifies the number of atoms in each block. Step size parameters for rigid
rotation, translation and cartesian moves are taken from the parameters of LIGMOVE.
- BINARY ntypea epsab epsbb sigmaab sigmabb: specifies a binary Lennard-Jones
system. ntypea is the number of type
A atoms--the rest are assumed to be type B and appear at the end of the list
of coordinates.
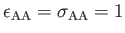 define the units of energy and length,
and epsab=
define the units of energy and length,
and epsab=
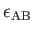 , epsbb=
, epsbb=
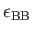 ,
sigmaab=
,
sigmaab=
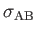 , sigmabb=
, sigmabb=
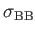 .
The box parameters and cutoff should be specified with the PERIODIC keyword.
.
The box parameters and cutoff should be specified with the PERIODIC keyword.
- BINSTRUCTURES SaveNth: requests that the geometry of every SaveNth
new structure found during basin-sampling is
recorded in binstructures.j, where j is the index of the bin
to which a given minimum belongs. If this keyword is
present then GMIN switches from plain PTMC to BSPT.
Without BINSTRUCTURES the BSPT keyword will perform a
standard PTMC run with no quenching.
- BLJCLUSTER ntypea epsab epsbb sigmaab sigmabb cutoff: specifies a binary Lennard-Jones
cluster. The parameters are the same as for BINARY, above.
- BLJCLUSTER_NOCUT ntypea epsab epsbb sigmaab sigmabb: specifies a binary Lennard-Jones
cluster without a distance cutoff. More efficient than BLJCLUSTER for smaller systems.
The parameters are the same as for BINARY, above.
[NB: Per-atom energies will be stored.]
- BLN
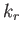
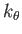 : specifies a BLN off-lattice protein model with
bond-length and bond-angle force constants and .
An auxiliary file BLNsequence is required.
See §6.2 for more details.
: specifies a BLN off-lattice protein model with
bond-length and bond-angle force constants and .
An auxiliary file BLNsequence is required.
See §6.2 for more details.
- BLNGO
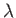 : specifies a Go potential
with the same form as the BLN potential. The parameters
and are the same as those used for the BLN keword and a BLNsequence file is required. Also needed is an auxiliary file, contactmap, containing the pairs of residues in contact in the native state of the protein
with one pair of residue numbers in each line of the file. An optional
parameter, , specifies the strength of the non-native interactions in a
scaled BLN potential [9].
: specifies a Go potential
with the same form as the BLN potential. The parameters
and are the same as those used for the BLN keword and a BLNsequence file is required. Also needed is an auxiliary file, contactmap, containing the pairs of residues in contact in the native state of the protein
with one pair of residue numbers in each line of the file. An optional
parameter, , specifies the strength of the non-native interactions in a
scaled BLN potential [9].
- BOXCENTROID x y z dx dy dz (ix iy iz): confine the system centroid to region (x
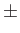 dx, ydy, zdz). Intended for systems interacting with an external field, e.g. a cluster supported on a substrate modelled using keyword MIE_FIELD. The cluster centroid will be checked after each random perturbation (before quenching!), and on each step of the QALCS_SURF procedure (if used, and also before quenching). When a centroid coordinate (say x_c) ventures outside the corresponding range (xdx), that centroid coordinate is translated back to x. If any of the optional integer parameters (ix, iy, iz) are set to unity (default value is zero), then the translation vector(s) in the corresponding direction(s) will be restricted to integer multiples of dx and/or dy and/or dz. This additional feature is intended for periodic external fields, e.g. a periodic substrate, in which case dx/dy/dz ought to match the period.
dx, ydy, zdz). Intended for systems interacting with an external field, e.g. a cluster supported on a substrate modelled using keyword MIE_FIELD. The cluster centroid will be checked after each random perturbation (before quenching!), and on each step of the QALCS_SURF procedure (if used, and also before quenching). When a centroid coordinate (say x_c) ventures outside the corresponding range (xdx), that centroid coordinate is translated back to x. If any of the optional integer parameters (ix, iy, iz) are set to unity (default value is zero), then the translation vector(s) in the corresponding direction(s) will be restricted to integer multiples of dx and/or dy and/or dz. This additional feature is intended for periodic external fields, e.g. a periodic substrate, in which case dx/dy/dz ought to match the period.
- BSMIN: specifies a Bulirsch-Stoer minimisation scheme.
Very inefficient compared to LBFGS.
- BSPT histmin histmax ptemin ptemax pttmin pttmax exchprob nequil ptsteps nquench nenrper hbins qfrq:
requests a basin-sampling run to accumulate the quench probability for local minima
as a function of potential energy using
a parallel-tempering algorithm.
This keyword also specifies the energy range for the histogram of quench energies,
histmin to histmax,
the energy range for the histogram of instantaneous configurations, ptemin to ptemax,
the temperature range (pttmin and pttmax),
the probability of attempting an exchange exchprob, the
number of equilibration steps, nequil,
the number of parallel tempering MC steps without quenching, ptsteps,
the number of parallel tempering MC steps with quenching, nquench,
the number of bins for the histogram of instantaneous potential energy, nenrper,
the number of bins for the histogram of quench energies, hbins,
and the quench frequency, qfrq.
Should be used together with the MPI keyword.
(This option is only available if the source is compiled with an MPI enabled.)
- BSPTDUMPFRQ n, n is the interval at which intermediate statistics
and bsptrestart files are dumped. If n is less than one these files
will only be dumped at the end of a complete run.
See also BSPTRESTART.
- BSPTRESTART: restart a previous BSPT or PTMC run.
The instantaneous and quench potential energy histograms are read from the last
Visits.his and Visits2.his files, and the current state from
bsptrestart files (one per node, numbered from zero).
A finished run can be continued with more steps by changing the nquench
or ptsteps parameters on the BSPT or PTMC line of
the data file. Setting the interval for BSPTDUMPFRQ to
minus one will read the last set of dump files.
- CALCQ: turn on calculation of bond order parameters.
- CAMSHIFT csversion svnDavid Wales shiftfile csn csalpha: uses chemical shifts as restraints during the
optimization procedure. Currently, CAMSHIFT can only be used together with CHARMM.
csversion is a string that specifies the method for combining the two potentials: MERGE means
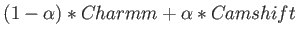 ,
ORIGINAL means
,
ORIGINAL means
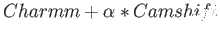 , and NOFF means
, and NOFF means
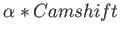 .
svnDavid Wales specifies the svn David Wales directory (e.g., $HOME/svn/). shiftfile is the file containing the
experimental shifts, which has to be located in the working directory. csversion, svnDavid Wales and shiftfile all have to be
specified together with CAMSHIFT. csn and csalpha are optional parameters. They define the
tolerance parameter of the CamShift energy profile, and the relation between CamShift and the force field, respectively.
Default values for both are 1.0.
.
svnDavid Wales specifies the svn David Wales directory (e.g., $HOME/svn/). shiftfile is the file containing the
experimental shifts, which has to be located in the working directory. csversion, svnDavid Wales and shiftfile all have to be
specified together with CAMSHIFT. csn and csalpha are optional parameters. They define the
tolerance parameter of the CamShift energy profile, and the relation between CamShift and the force field, respectively.
Default values for both are 1.0.
- CAPSID rho epsilon radius height: specifies a coarse-grained potential to represent virus capsid pentamers
with parameters
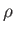 ,
,
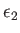 ,
, 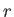 and
and 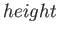 , respectively.
If is omitted the default is 0.5.
, respectively.
If is omitted the default is 0.5.
- CENTRE : if present the system will be translated so that the centre-of-mass
lies at the origin after every quench.
- CENTREXY : if present the system will be translated so that the centre-of-mass
lies at the centre of the xy plane after every quench. This is useful when using an implicit membrane like IMM1 where you have directionality only in the
z-direction, so centreing in x and y should have no delaterious effect.
- CG : specifies a conjugate-gradient minimisation scheme. Inefficient compared to LBFGS.
- CHANGEACCEPT naccept: naccept is an integer which sets the interval
at which the acceptance ratio is checked and possible adjustments are made to the maximum
step size or the temperature. The default is naccept
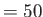 .
.
- CHARMM: specifies that a CHARMM potential should be used.
See also keywords CHARMMTYPE, CHPMAX, CHPMIN, CHNMAX, CHNMIN,
NOPHIPSI, TOMEGA, INTMIN, CHFREQ, CHRIGIDROT,
CHRIGIDTRANS, and RMS. If CHNMAX is not specified, a cartesian
displacement step taking scheme will be used. For cartesian steps, rings are moved as rigid bodies to avoid false knotted minima. See RINGROTSCALE. Finally, Molecular Dynamics (MD) can be employed to generate new geometries. See CHMD
- CHARMMDFTB: specifies that the CHARMM SCC-DFTB potential is to be used, and
disables updates of the nonbonded list. This assumes you are using a fully QM system. If you
are using a QM/MM setup, you should not use this keyword! Note that SCC-DFTB can only be used
with CHARMM35.
- CHMD CHMDFREQ: Requests Molecular Dynamics (MD) runs to be performed every CHMDFREQ step to generate new geometries. A CHMDFREQ setting of 20 will execute an MD run every 20
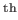 step, while dihedral or cartesian moves are applied otherwise as specified in the data file. A CHARMM parameter file named 'chmd.par' containing all relevant keywords for the CHARMM DYNA module has to be present in the working directory. All CHARMM keywords must be uppercase and given in the first line. A typical example is:
step, while dihedral or cartesian moves are applied otherwise as specified in the data file. A CHARMM parameter file named 'chmd.par' containing all relevant keywords for the CHARMM DYNA module has to be present in the working directory. All CHARMM keywords must be uppercase and given in the first line. A typical example is:
VERL NSTEP 500 TIMESTEP 0.002 TWINDH 10.0 IEQFRQ 200 ICHECW 1 IASORS 0 IASVEL 1 FIRS 500 FINA 500
Please consult the CHARMM manual for further details on the DYNA module. Currently, the length of the input string given in 'chmd.par' is limited to 500 characters.
- CHARMMENERGIES: prints the components of the total CHARMM energy after each step.
- CHARMMTYPE topfile paramfile: topfile and paramfile are the
common CHARMM top and param files , e.g., `toph19_eef1_perm.inp' and `param19_eef1_perm.inp'.
- CHFREQ nfreq: used with CHARMM keyword to specify that every
nfreq basin-hopping steps dihedrals are twisted. Default is nfreq=1.
- CHNMAX: used with CHARMM keyword to specify the maximum allowed
number of angles to be twisted. Specifies a dihedral angle step taking scheme.
- CHNMIN: used with CHARMM keyword to specify the minimum allowed
number of angles to be twisted.
- CHPMAX: used with CHARMM keyword to specify the maximum allowed
probability for twisting an angle.
- CHPMIN: used with CHARMM keyword to specify the minimum allowed
probability for twisting an angle.
- CHRIGIDROT prot rotmax nrot: used with CHARMM keyword
to support rigid body rotation every nrot basin-hopping steps with maximum allowed
probability prot and maximum allowed rotation angle rotmax (in degrees).
The keyword CHRIGIDROT requires a file segments.tomove, which specifies
the segments for rigid rotation. The segments are numbered and each line contains only one number.
- CHRIGIDTRANS ptrans transmax ntrans: used with CHARMM keyword
to support rigid body translation every ntrans basin-hopping steps with maximum allowed
probability ptrans and maximum allowed translation transmax (in Å).
The keyword CHRIGIDTRANS requires a file segments.tomove, which specifies
the segments for rigid translation. The segments are numbered and each line contains only one number.
CHRIGIDROT and CHRIGIDTRANS use the same segments.tomove.
- CHECKD n: calculates gradients analytically and numerically for the initial coordinates, then exits.
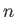 is an
integer optional argument; if equal to zero, then only the single-point energy is calculated and reported.
is an
integer optional argument; if equal to zero, then only the single-point energy is calculated and reported.
- CISTRANS: disables all checks for cis or deformed amide/peptide bonds.
- CHIRO
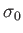
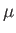
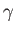
![$ [L]$](img63.png) : calls the OK potential. If
: calls the OK potential. If 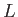 is absent or 0, then use one LJ site. If
is absent or 0, then use one LJ site. If 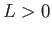 , use LJ-like rods of length
, use LJ-like rods of length 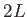 . Output is written to chiro.xyz. A periodic boundary condition in the
. Output is written to chiro.xyz. A periodic boundary condition in the 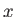 direction can be applied using the PERIODIC keyword.
direction can be applied using the PERIODIC keyword.
- COLDFUSION thresh: if the energy falls below threshold thresh then
cold fusion is assumed to have occurred and geometry optimisation stops.
The default value is
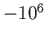 .
.
- COMPRESS kcomp: add a harmonic compression potential with force constant kcomp using the
centre-of-mass distance for each atom.
- COMPRESSRIGID kcomp dist: if at least one rigid body's centre of mass is further than dist from its nearest neighbour,
add a harmonic compression potential with force constant kcomp using the centre-of-mass distance for each rigid body. The compression is
disabled once the rigid bodies are all within dist of another.
- COMMENT : the rest of the line is ignored.
- COOPMOVE n cut: specifies cooperative moves in the step-taking routine. An atom is
selected at random, and the n nearest neighbours (default 5) that lie within a cutoff
distance of cut (default 1.0) are moved by the same amount.
- CPMD sys: specifies that the CPMD program should be called for energies and gradients. Not
tested!
- CUTOFF cutoff: sets a cutoff beyond which the potential is truncated. This
only has an effect for tight-binding silicon at present. Interaction cutoffs for other potentials
should be specified in their input files e.g. min.in (and min_md.in if used)
for AMBER9 or below the CHARMM line for CHARMM.
- DBP epsbb sigmabb muD E: calls
a finite system of dipolar Lennard-Jones dumbbells [10], where the electric field of stength
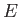 can be
optionally present. The field, if present, is along the space-fixed z-direction.
can be
optionally present. The field, if present, is along the space-fixed z-direction.
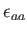 and
and
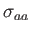 are both set to unity by default.
are both set to unity by default. 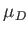 is the dipole moment;
is the dipole moment; 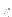 's
and
's
and 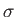 's correspond to the Lennard-Jones parameters.
's correspond to the Lennard-Jones parameters.
- DBRENT: specifies minimisation using Brent's method with first derivatives in the
conjugate-gradient procedure.
Inefficient compared to LBFGS.
- DEBUG: sets various debug printing options including the dumping of initial
geometries and energies (to dump.X.xyz) if DUMP is also set.
- DECAY x: magnitude of random move decays according to parameter
x with distance from a randomly chosen atom.
- DF1: specifies a binary 2D potential.
The first
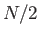 atoms have unit radius and the rest
have radius 1.4, with a cutoff for each pair type at the
average radius.
The keyword 2D must also be specified, along with a
PERIODIC line to specify two box-lengths.
Initial work uses box lengths of 3.31437171 for a number density of 0.9.
atoms have unit radius and the rest
have radius 1.4, with a cutoff for each pair type at the
average radius.
The keyword 2D must also be specified, along with a
PERIODIC line to specify two box-lengths.
Initial work uses box lengths of 3.31437171 for a number density of 0.9.
- DFTB: specifies a DFT-based tight-binding potential; the multiplicity is specified by
keyword MULTIPLICITY.
- DFTBC: specifies a DFT-based tight-binding potential carbon.
Can be guided using LJAT.
- DGUESS dguess: initial guess for diagonal elements of the inverse
Hessian, used whenever the LBFGS optimiser is reset.
The default is dguess=0.1.
- DIHEDRALROTATION: This keyword performes random rotations of groups about specified dihedral angles. Similar to GROUPROTATION, the dihedral groups must be defined in a file named dihedralgroups, with the following syntax:
GROUP name type at1 at2 at3 at4 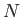 maxrot pselect
maxrot pselect
gat
gat
gat
Here ``name'' is a string defining the group name; ``type" is a string defining the type of dihedral for backbone rotations (phi, psi etc.), used for printing purposes only; at1 to at4 are integers specifying the atom indices defining the dihedral angle, gat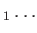 gat define indices of the atoms which rotate, and the number of such atoms.
gat define indices of the atoms which rotate, and the number of such atoms.
Note: at4 should also be included in the list of atoms to rotate, as it is the final atom defining the dihedral!
- DIPOLES: causes the first order induction energy to be included
in a diatomics-in-molecules calculation for Ne
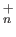 or Ar. By default this
term is neglected, although it may be significant.
or Ar. By default this
term is neglected, although it may be significant.
- DONTMOVE n1 n2 : prevents atoms n1, n2, moving during MC step taking. They can still move during minimisation.
- DONTMOVEGROUP centre radius type: If type is set as default to GT, DONTMOVEGROUP prevent all atom greater than radius angstroms from the centre atom from moving during MC step taking. type can also be set to LT to not move all atoms within radius of centre.
- DONTMOVERES n1 n2 : prevents all atoms in residues n1, n2, from moving during MC step taking.
- DONTMOVEALL n1 n2 : prevents all atoms from moving during MC step taking (usually used with DOMOVE and DOMOVERES).
- DOMOVE n1 n2 : allows atoms n1, n2, to move during MC step taking. Only functions in conjunction with
DONTMOVEALL
- DOMOVERES n1 n2 : allows all atoms in residues n1, n2, to move during MC step taking. Only functions in conjunction
with DONTMOVEALL
- DUMP: if present will cause the energy and quench geometry for every step
to be dumped into dump.X.xyz where X is an integer. The geometries are saved
in XMakemol xyz format. If CHARMM is also specified, dump.pdb and dump.crd
are produced containing each quench geometry in PDB and CHARMM CRD format.
- DUMPINT int: changes the default interval for dumping a restart
GMIN.dump file from 1000 basin-hopping steps to int.
- DUMPMIN: When using more than one processor (e.g. BHPT), or in conjunction with DUMPSTRUCTURES, the SAVE lowest minima will be dumped every
DUMPINT steps as dumpmin.x files.
- DUMPQU: when using AMBER9, dumps each quench geometry in rst format to quenchX.rst
and pdb format to quenchX.pdb. Dumping does not occur if a chirality check fails.
- DUMPSTEPS: when using AMBER9, dumps each geometry after the MC step has been taken in rst format to afterstepX.rst
and pdb format to afterstepX.pdb.
- DZUGUTOV dzp1 dzp2 dzp3 dzp4 dzp5 dzp6 dzp7: Dzugutov potential in a general form.
The parameters are
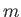 ,
, 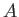 ,
, 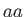 ,
,  ,
,  ,
,  and
and 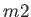 .
.
- EAMAL: specifies an embedded atom model for aluminium.
- EAMLJ A0 beta Z0: specifies the EAMLJ potential (Baskes, Phys. Rev. Lett.,
27, 2592, 1999) with parameters A0, beta and Z0.
- EDIFF econv: quench minima are only considered to be different if their
energies differ by at least
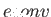 . This option mainly affects the lowest energy
saved geometries. If the current quench energy is within of a saved energy, but
lies lower, then the saved energy and geometry are replaced.
The default is
. This option mainly affects the lowest energy
saved geometries. If the current quench energy is within of a saved energy, but
lies lower, then the saved energy and geometry are replaced.
The default is 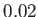 but different values are appropriate for different potentials.
but different values are appropriate for different potentials.
- ENPERMS: Enumerate all distinct permutations of a binary system (0 < NTYPEA < NATOMS) using Lehmer's algorithm. Instead of a traditional geometry-perturbing step, Lehmer's algorithm will generate a new permutationl isomer by swapping the coordinates of two deterministically-picked unlike atoms. The procedure terminates once all the distinct permutations have been enumerated.
- EQUILIBRATION equil DumpEveryNthQuench: equil is the number of
MC steps preceding the accumulation of the
density of states histogram in a Wang-Landau
basin-sampling run. The default is 0. DumpEveryNthQuench specifies how often the
statistics are recorded into the output files.
- EXEQ n: for use with the RESERVOIR keyword. After a configuration is
taken from the reservoir by the lowest temperature non-reservoir replica we do not
record visits statistics for n steps. This allows some local equilibration if
the configuration is not truely equilibrium.
- EXPANDRIGID freq factor (NORMALISE): for use with the generalised rigid body framework,
RIGIDINIT. Expands the system by a factor of factor by scaling the distance
of each rigid body from the centre of mass of the system every freq steps. If the third argument NORMALISE
is used, all rigid bodies are simply translated by factor from the system centre of mass.
- FAL : specifies the Farkas potential for aluminium.
- FEBH
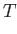 FEBH: do a free energy basin-hopping run. This uses the free energy
calculated using the harmonic superposition approximation for the acceptance criterion. This also
leads to rejection of transition states from the Markov chain (GMIN has no in-built check to guarantee
this for a potential energy run). Additionally, it enables the MIN_ZERO_SEP keyword, which
uses the separation of the zero eigenvalues (typically the 6 corresponding to overall translation and
rotation) to determine convergence.
FEBH: do a free energy basin-hopping run. This uses the free energy
calculated using the harmonic superposition approximation for the acceptance criterion. This also
leads to rejection of transition states from the Markov chain (GMIN has no in-built check to guarantee
this for a potential energy run). Additionally, it enables the MIN_ZERO_SEP keyword, which
uses the separation of the zero eigenvalues (typically the 6 corresponding to overall translation and
rotation) to determine convergence.
- FIXEDEND : requires documentation.
- FIXBOTH : both the temperature and maximum step size are fixed regardless of
the calculated acceptance ratio.
- FIXCOM : fix centre of mass rather than centre of coordinates.
- FIXSTEP : the maximum step size is fixed and the temperature is varied to
try and achieve the requested acceptance ratio.
- FIXTEMP : explicitly fixes the temperature. Only used if FIXSTEP is set, in
which case using FIXTEMP gives a result equivalent to FIXBOTH.
- FNI : specifies the Farkas potential for nickel.
- FRAUSI : specifies a particular tight-binding potential for silicon.
See also keyword ANGSTROM.
- FREEZE n1 n2 : freeze the coordinates of atoms n1, n2,. Atoms affected by FREEZE will not move during MC step taking,
or during minimisation as their gradients are set to zero.
- FREEZEGROUP centre radius type: If type is set as default to GT, FREEZEGROUP FREEZEs all atom greater than radius angstroms from the centre atom. type can also be set to LT to FREEZE all atoms within radius of centre.
- FREEZERES n1 n2 : freeze the coordinates of all atoms in residues n1, n2,.
- FREEZEALL n1 n2 : freeze the coordinates of all atoms
- UNFREEZE n1 n2 : unfreeze the coordinates of atoms n1, n2,. Only functions in conjunction with
FREEZEALL
- UNFREEZERES n1 n2 : unfreeze the coordinates of all atoms in residues n1, n2,. Only functions in conjunction with
FREEZEALL
- FS gatom: specifies a Finnis-Sinclair potential using parameters from
Finnis and Sinclair, Phil. Mag. A, 50, 45 (1984)
and corresponding erratum Phil. Mag. A, 53, 161 (1986).
gatom=1 for V, gatom=2 for Nb, gatom=3 for Ta, gatom=4
for Cr, gatom=5 for Mo, gatom=6 for W, gatom=7
for Fe (original parameters), gatom=8 for Fe (modified parameters in erratum).
Subtoutine FS was coded by Ja,es Elliott in April 2009.
- GA population offspring generations: specifies optimisation using a
``Lamarckian'' genetic algorithm[7] instead of Monte Carlo. If
STEPS is defined, each new member of the population is optimised with a
basin-hopping Monte Carlo search. The search will end when the number
of generations has been exhausted or when solution specified by
the TARGET keyword has been reached. Currently, this is only implemented
for the BLN model protein and clusters. See §3.2 for more
details.
- GABHINCR increment: specifies the use of a hybrid
GA/basin-hopping algorithm with a variable length of basin-hopping search. The
increment defines the number of extra basin-hopping steps to perform in
each generation. This does not have to be an integer (e.g. increment =
0.5 increases the number of basin-hopping steps by one after every other
generation).
- GACROSS n: specifies n-point crossover for genetic algorithm
mating operations. Permitted values for n are 1 and 2 (default = 1).
- GADUPPRED ethresh: sets the energy threshold for the GA
duplicate predator. If two structures differ by less than ethresh, they are marked as duplicates and the least stable one is
removed from the population.
- GAEPOCH ethresh save DUMP: specifies the use of a new epoch
operator to restart GA searches that have converged. If the mean energy of
the population decreases by less than ethresh in one generation, a new
population of random solutions is generated and a new epoch begins. Optionally,
the best save solutions are carried over from one epoch to the next.
Adding the optional DUMP statement prints the conents of the population to
a file epoch.n at the end of each epoch.
- GAINITCHAIN: random structures at the start of an epoch
in a GA search will be generated as a sequence continuous chain. If
no GAINIT... option is specified, this will be chosen for BLN
proteins.
- GAINITSPHERE radius: random structures at the start of an epoch
in a GA search will be generated as points inside a sphere. If
no GAINIT... keyword is specified, this will be chosen for clusters.
The default radius is 3.0.
- GAMUTRATE mutrate: sets the mutation rate for GA
searches. Default=0.1.
- GASELROUL tournsize: specifies the roulette wheel method to select
parents for mating in GA searches. The probabilities for the roulette
wheel are generated using a tanh fitness function.
- GASELTOURN tournsize: specifies the tournament method to select
parents for mating in GA searches. Default tournsize=3.
- GBD kappa kappaprime mu nu sigma0 epsilon0: calls a finite system of discoids
interacting via a modified Gay-Berne pair potential due to Bates and Luckhurst [11].
- GCBH mu nmax int relax prob: specifies grand canonical basin-hopping.
mu is the chemical potential, nmax is the maximum number of atoms,
int is the interval between changes in the number of atoms,
relax is the block size for relaxation with conventional BH steps,
prob is the probability of increasing the number of atoms by one,
and 1-prob is the probability of removing an atom.
The grand canonical accept/reject is based on the lowest potential obtained within
a Markov chain of blocks, to allow the system to relax when the number of atoms
increases or decreases.
The atom removed is currently programmed to be the one that is most weakly bound, so
this pair energy must be known.
A new atom is added two distance units outside the current cluster defined
by the atom furthest from the centre of coordinates.
In both cases an initial quench is performed immediately, and the attempt is rejected
if this quench is unsuccessful.
Some declarations are based on nmax, while locally dynamic allocations
for subroutines can use the current number of atoms.
If FEBH is specified then the potential is based on the exponential factor
and the partition function (including rotation if USEROT is used).
Otherwise, the test is based on the potential defined by
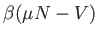 .
.
- GLJ n1 n2 n3 etc.: specifies general Lennard-Jones potential for any number of
differ LJ species.
The and parameters for all the different species should be specified as
the lower diagonal blocks of the corresponding matrices on the following lines.
For example, to specify a 13-atom cluster with three different species:
GLJ 6 4 3
1.0
1.1 1.2
1.2 1.3 1.4
1.0
1.5 1.6
1.7 1.8 1.9
- GLJY alpha A xi: specifies a combination of generalised Lennard-Jones and Yukawa potentials for colloidal systems.
The potential is given by
![$ U(r) = 4\epsilon \left[ (\sigma/r)^{2\alpha} - (\sigma/r)^{\alpha}) \right] + A \exp(-r/\xi) / (r/\xi)$](img93.png) in reduced units with
in reduced units with
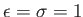 .
.
- GROUND: when combined with keywords NEON or ARGON
uses an accurate (Aziz) potential to model the ground state neutral cluster.
- GROUPROTATION (freq) (scalemode) (offset): specifies group rotation moves for groups of atoms defined in atomgroups. freq
(optional) specified the frequency with which these moves should be made. The default, 1, specified group rotations be made every 1 steps.
scalemode (optional) can be used to determine how the GROUPROTATION moves are scales to attempt to achieve the desired acceptance ratio.
There are 4 options for scalemode - SCALENONE (default): do no scaling, SCALEPROB: scale the group selection probabilities, SCALEROT: scale
the rotation amplitudes and SCALEBOTH: scale both selection probabilities and rotation amplitudes.
offset (optional) can be used for systems with consistant ligand or cofactors to allow the ligand/cofactor group numbering to be
system independant. For example, if there are 3500 atoms in a protein, and the ligand starts at atom 3501, setting offset to 3500 means
that the GROUP in atomgroups numbering starts at 1 again. The default offset is 0. Currently this is only usable with
AMBER9 and CHARMM.
The atomgroups file is formatted as follows:
GROUP name bondatom1 bondatom2 groupsize rotationscalefactor probselect
groupatom1
groupatom2
groupatom3
...
The group rotation axis is defined by the vector from bondatom1->bondatom2, and the rotation is scaled by rotationscalefactor
.
Here is an example atomgroups file containing two groups:
GROUP OME 6 5 4 1.0 0.8
1
2
3
4
GROUP CH2OH 23 25 4 1.0 0.8
26
27
28
29
- QUIETGROUPROT: suppresses output from group rotation about which angles were changed.
- GTHOMSON type param1 param2 param3 param4: the system will be constrained to a curved surface.
The type of surface is specified by type as follows:
1 A cylinder, with height param1 and radius param2.
2 A catenoid
3 An unduloid
4 An unduloid
5 A sphere, with radius param1.
6 A Möbius strip, with radius param1 and width param2.
- GTHOMSONPOT type : specifies the potential to use with the surface described by
the GTHOMSON keyword. The values of type give the following behaviours:
1 A Coulomb potential, with all particles having a charge of 1.
2 A
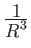 potential.
potential.
3 A Yukawa potential, with screening length .
4 A Lennard-Jones potential, with characteristic distance and a well depth of 1.
5 A repulsive Lennard-Jones (
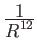 ) potential,
with characteristic distance .
) potential,
with characteristic distance .
6 A Morse potential, with characteristic distance and range parameter .
- GUIDE guidecut: specifies the RMS force below which the real potential is used
rather than a guiding potential. The systems affected are CPMD and WELCH,
which are guided by AMBER and TOSI, respectively, and also PACHECO,
where the Axilrod-Teller contribution is only included when the RMS force falls below
guidecut. Default guidecut=0.0001.
New guided potentials are ZETT1 and ZETT2 (guided by Morse with
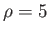 ) and
NATB (guided by GUPTAT). Parameters for the guiding potential must also be specified in
data.
) and
NATB (guided by GUPTAT). Parameters for the guiding potential must also be specified in
data.
- GUPTA gatom: specifies a Gupta potential using parameters from Cleri and Rosato,
Phys. Rev. B, 48, 22 (1993). gatom=1 for Ni,
gatom=2 for Cu,
gatom=3 for Rh,
gatom=4 for Pd,
gatom=5 for Ag,
gatom=6 for Ir,
gatom=7 for Pt,
gatom=8 for Au,
gatom=9 for Al,
gatom=10 for Pb,
gatom=11 for Ti type 1,
gatom=12 for Ti type 2,
gatom=13 for Zr type 1,
gatom=14 for Zr type 2,
gatom=15 for Co,
gatom=16 for Cd type 1,
gatom=17 for Cd type 2,
gatom=18 for Zn,
gatom=19 for Mg,
gatom=20 for V,
gatom=21 for Na,
gatom=22 for Sr (Wang and Blaisten-Barojas, J. Chem. Phys., 115, 3640 (2001)),
gatom=22 for Au as used by Garzon et al.
The Gupta subroutine was recoded more efficiently by James Elliott in April 2009.
- HBONDLIGAND (ligandresn): used with the HBONDMATRIX keyword. When specified, groups are defined only
by the hydrogen-bonds between the ligand and protein only - not between sidechains. The optional arguement ligandresn
specifies which line in the residuefile corresponds to the ligand. The default is to assume it is the last line of the file.
This keyword must be specified after HBONDMATRIX in the data file.
- HBONDMATRIX donoracceptorfile residuefile mode: used with the AMBER keyword to focus GMIN sampling on
the diversity of ligand binding-modes in a protein/ligand system. donoracceptorfile should contain the AMBER ptraj
definitions of the hydrogen-bond donor and acceptor atoms for your system as described in the AMBER Tools manual (see Google).
residuefile should contain the list of residue numbers (one per line) that are involved in binding. When you have no
specific information, this can just mean those in close proximity to the binding site. mode is an optional arguement, which
defaults to ACCEPT. If REJECT is used instead, HBONDMATRIX will instead restrict sampling to the binding mode identified after
the initial quench. This can be used to optimise a single binding mode.
- HBONDSOFTCUT dloose dtight aloose atight: used with HBONDMATRIX to specify a custom cutoff window for the
hydrogen-bond analysis used by the HBONDMATRIX keyword to group structures. The default values for the four parameters are
3.05, 2.95, 115.0 and 120.0. It should be noted that the actual angles used in the analysis are
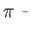 aloose and
atight. This is why the looser angle cutoff is actually numerically smaller.
aloose and
atight. This is why the looser angle cutoff is actually numerically smaller.
- HOMOREF smode gmode ncycles kmax: performs homotop refinement for a binary system (with NTYPEA between 0 and NATOMS) using an iterated local search (ILS). The refinement happens every basin-hopping step, after the coordinates have been perturbed and quenched. It involves exchanging the coordinates of two unlike atoms until a termination condition is met. The termination conditions (and other specifics) depend on smode: smode = 0 is steepest-descent-like, smode = 1 is strictly downhill with a memory effect, and smode = 2 is akin to the Kernighan-Lin procedure that can also climb uphill. gmode specifies the method for forecasting flip gains: gmode = 2 is brute-force enumeration of all swap gains on each step; gmode = 1 utilises a sequence of flips with each flip gain evaluated exactly; and gmode = 0 is also based on a sequence of flips, with each flip gain approximated to achieve more efficient but less reliable results. The maximum length of the flip sequence is specified by kmax. Finally, ncycles specifies the desired number of search restarts (i.e. cycles), and, if ncycles exceeds one, then a random permutation operator will be used to perturb the best solution encountered so far, with the result used to initialise the next cycle.
- HOMOREFTEST: prints extra info for testing HOMOREF.
- HOMOREFCHECK: specifies that a converged sequence of flips during homotop refinement is to be checked by evaluating all the swap gains for the final solution. The check is passed only if all the swap gains are non-negative. (This is intended for HOMOREF with gmode set to 0 or 1.)
- HOMOREF_AUX nswaps temp factor nncut : specifies that an auxiliary basin-hopping run with nswaps exchanges is to be performed after each random permutation step during homotop refinement (see HOMOREF). The basin-hopping procedure is rejection-free, with each atom
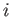 weighted by a Boltzmann factor
weighted by a Boltzmann factor 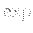 (
(
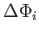 /temp), where temp behaves like temperature, and
/temp), where temp behaves like temperature, and
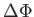 is a change in an auxiliary bond-counting potential constructed from and averaged over the previously encountered locally optimal homotops. factor is a multiplicative factor used to update the temperature after each exchange. nncut is the nearest neighbour cutoff distance used in the construction of the auxiliary bond-counting potential.
is a change in an auxiliary bond-counting potential constructed from and averaged over the previously encountered locally optimal homotops. factor is a multiplicative factor used to update the temperature after each exchange. nncut is the nearest neighbour cutoff distance used in the construction of the auxiliary bond-counting potential.
- HYBRIDMIN rigidconv: enables hybrid rigid body/all-atom minimisation when using RIGIDINIT. rigidconv is
the RMS force convergence criterion for the rigid body minimisation. Once converged, an all-atom minimisation begins using the
convergence specified in SLOPPYCONV. The final quenches are done atomistically.
- INTMIN: used with CHARMM keyword to specify minimisation in internal
coordinates. This generally appears to be
slower than using Cartesian coordinates.
- INVERTP: specifies the inverted potential. The energy and all derivatives
are multiplied by
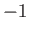 .
This converts searches for the highest index saddles into minimisation.
However, it is only likely to work for bounded potentials.
.
This converts searches for the highest index saddles into minimisation.
However, it is only likely to work for bounded potentials.
- JC: Specifies Murrell's two- plus three-body
potential.[12,13,14,15,16]
A file JMparams must
exist in the current directory containing the parameters
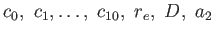 and
and 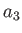 . An optional cutoff parameter can also be provided at the end of the
JMparams file.
Subroutines used: jmec, jm2c, jm3c.
. An optional cutoff parameter can also be provided at the end of the
JMparams file.
Subroutines used: jmec, jm2c, jm3c.
- JUMPMOVE np1 np2 int: specify J-walking type attempts between parallel runs np2
and np1 at intervals of int steps.
- LB2 specifies the potential[17,18,19]
![$\displaystyle V = \frac{\epsilon}{2} \sum_{i<j} \left[ \left(\frac{r_{ij}}{\sigma}\right)^2+ \left(\frac{\sigma}{r_{ij}}\right)^2 \right],$](img105.png) |
(1) |
where and are set to unity.
- LFLIPS n m kT (mfac): for every nth step in the main basin-hopping sequence perform a subsequence of m steps with flip moves only. This subsequence constitutes an independent block of semi-grand canonical basin-hopping at a given kT, with the total number of atoms fixed but the relative population of constituent species allowed to vary. In every instance the subsequence starts with the specified value of kT, which is then multiplied by the (optional) parameter mfac on each of the m successive steps. (Default: mfac = 1.) The keyword SEMIGRAND_MU can be used to impose non-zero semi-grand chemical potentials (with respect to the first species).
- LFLIPS_RESET: at the start of every subsequence of flips, the stoichiometry will be reset to the value inferred from the keyword specifying the multicomponent potential. The atomic labels will be reassigned randomly.
- LIGMOVE ligrotscale ligcartstep ligtransstep ligmovefreq: used with AMBER9 and MOVABLEATOMS. Specifies ligand only rotation, cartesian perturbation and translation. The ligand is defined by atom index in the file 'movableatoms'. Setting ligrotscale less than 1.0
limits the ammount of rotation possible - this may be required to prevent cold fusion with non-spherical ligands. ligcartstep and ligtransstep
define the maximum size (in angstroms) of the random cartesian perturbations and rigid body translation applied to the ligand respectively.
ligmovefreq can be set to greater than 1 to prevent ligand moves being applied every step i.e. ligmovefreq = 2 for every other step.
All ligand moves are applied AFTER any MD if AMBERMDMOVES is on to prevent the MD exploding.
- LJAT
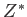 scale: specifies the Lennard-Jones plus Axilrod-Teller
potential for a reduced triple-dipole struength of .
Provided mostly as a guiding function for DFTBC.
rescale is the coordinate rescaling factor from the LJAT potential
to DFTB, default value 2.424.
scale: specifies the Lennard-Jones plus Axilrod-Teller
potential for a reduced triple-dipole struength of .
Provided mostly as a guiding function for DFTBC.
rescale is the coordinate rescaling factor from the LJAT potential
to DFTB, default value 2.424.
- LJCOUL nc
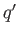 f
f
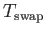 specifies a cluster of Lennard-Jones particles in which the first nc
particles carry identical reduced charges in addition to the Lennard-Jones interaction.
The parameter f specifies what fraction of the Monte Carlo steps should be swaps between the
positions of a charged and a neutral particle, rather than a conventional step.
is the temperature to be used in the acceptance criterion for swap moves, overriding
that specified using the TEMPERATURE keyword. Generally, a lower temperature is more effective
at finding the lowest-energy permutation of charges. The default value of
is zero.
The reduced charge is related to the actual charge
specifies a cluster of Lennard-Jones particles in which the first nc
particles carry identical reduced charges in addition to the Lennard-Jones interaction.
The parameter f specifies what fraction of the Monte Carlo steps should be swaps between the
positions of a charged and a neutral particle, rather than a conventional step.
is the temperature to be used in the acceptance criterion for swap moves, overriding
that specified using the TEMPERATURE keyword. Generally, a lower temperature is more effective
at finding the lowest-energy permutation of charges. The default value of
is zero.
The reduced charge is related to the actual charge 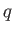 by
by
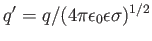 ,
where and are the Lennard-Jones well depth and length parameter respectively.
This way, the reduced energy of two charges is
,
where and are the Lennard-Jones well depth and length parameter respectively.
This way, the reduced energy of two charges is
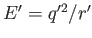 , where
, where
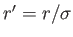 is the reduced distance
bdetween the charges.
is the reduced distance
bdetween the charges.
- LOCALSAMPLE abthresh acthresh: Keyword currently under construction! For three groups of atoms defined in movableatoms
(A,B,C), a step is quenched when the centre of coordinates of A->B is less than abthresh AND A->C is less than acthresh.
If this condition is broken AFTER the quench, it is automatically rejected.
- LSWAPS n m kT (mfac): for every nth step in the main basin-hopping sequence perform a subsequence of m steps with exchange moves only. The keyword is intended for homotop optimisation at regular intervals for a given kT. In every instance the subsequence starts with the specified value of kT, and this value is multiplied by the (optional) parameter mfac on each of the m successive steps. (Default: mfac = 1.)
- MAKEOLIGO START
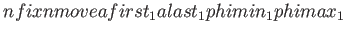 dmin dmax SCONLY
specifies the oligomer generation procedure. Here, the sample input follows for the generation of a dimer.
The argument START specifies that a new oligomer is to be generated.
The second and third arguments determine how many peptide chains are fixed (nfix) and relocatable (nmove), respectively.
The input geometry has
to be provided in such a form that all fixed peptide chains come first, followed by the peptide chains,
which are set to
new positions during the oligomer generation procedure. The secondary structures of the relocatable peptides are determined via the input
geometry. The following 4
dmin dmax SCONLY
specifies the oligomer generation procedure. Here, the sample input follows for the generation of a dimer.
The argument START specifies that a new oligomer is to be generated.
The second and third arguments determine how many peptide chains are fixed (nfix) and relocatable (nmove), respectively.
The input geometry has
to be provided in such a form that all fixed peptide chains come first, followed by the peptide chains,
which are set to
new positions during the oligomer generation procedure. The secondary structures of the relocatable peptides are determined via the input
geometry. The following 4 nmove arguments specify the first and last atom for each of the relocatable peptide chains, and the
minimum and maximum angle between which the relocatable peptide in question is to be positioned in the
nmove arguments specify the first and last atom for each of the relocatable peptide chains, and the
minimum and maximum angle between which the relocatable peptide in question is to be positioned in the 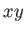 -plane:
-plane:
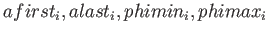 with
with
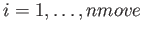 .
The next two arguments determine the minimum and maximum distances, dmin and dmax, which define the boundaries
for the relocation of the peptide chains with respect to the centre of mass of the fixed part of the input structure.
The effect of the last argument SCONLY is that during the subsequent optimisation of the oligomer only the dihedral angles
of the sidechains are perturbed. If this argument is not present, the backbone dihedrals are also changed.
The MAKEOLIGO keyword also affects the rigid body translation and rotation during the subsequent optimisation. The translation is
only performed in the -plane. The rotation can be performed either around the
.
The next two arguments determine the minimum and maximum distances, dmin and dmax, which define the boundaries
for the relocation of the peptide chains with respect to the centre of mass of the fixed part of the input structure.
The effect of the last argument SCONLY is that during the subsequent optimisation of the oligomer only the dihedral angles
of the sidechains are perturbed. If this argument is not present, the backbone dihedrals are also changed.
The MAKEOLIGO keyword also affects the rigid body translation and rotation during the subsequent optimisation. The translation is
only performed in the -plane. The rotation can be performed either around the 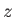 -axis only or in the in the full three-dimensional
space, depending on whether the first argument is START (as in this example) or INITROT. The arguments following
INITROT are identical to those following START.
-axis only or in the in the full three-dimensional
space, depending on whether the first argument is START (as in this example) or INITROT. The arguments following
INITROT are identical to those following START.
- MAXBFGS max: max is the largest permitted LBFGS step.
- MAXERISE maxez: specifies the largest rise in energy permitted during an LBFGS
minimisation. MAXERISE must to be large enough for discontinuities encountered
during quenches to be ignored, or the quench may fail. The default is
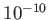 , or
, or 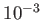 , for periodic boundary conditions, where
discontinuities can arrise due to multiple images.
, for periodic boundary conditions, where
discontinuities can arrise due to multiple images.
- MAXIT maxit maxit2: maxit and maxit2 are integers specifying the
maximum number of iterations allowed in the conjugate gradient quenches. maxit applies
to the `sloppy' quenches of the basin-hopping run and maxit2 to the final quenches
that are used to produce the output in file lowest.
- MGGLUE: specifies a glue potential for magnesium
- MLOWEST: Like TARGET. Accepts multiple target energies and will stop upon hitting *all* targets as opposed to *any* target.
- MORSE rho: specifies a Morse potential
with range parameter rho.[20,21,22]
- MPI: specifies an MPI parallel job.
(only available if the source is compiled with MPI enabled).
- MGUPTA nspecs A_11 p_11 q_11 xi_11 r0_11: specifies a multicomponent Gupta potential for nspecs distinct metallic species. If nspecs = 1 then this single line is sufficient. The model parameters A_11, p_11, q_11, xi_11 and r0_11 have the same meaning as for the keyword BGUPTAT. If nspecs
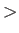 1, then
1, then
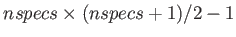 more subsequent lines of the form MGUPTA (n_I) A_IJ p_IJ q_IJ xi_IJ r0_IJ must be supplied for all the remaining I,J
more subsequent lines of the form MGUPTA (n_I) A_IJ p_IJ q_IJ xi_IJ r0_IJ must be supplied for all the remaining I,J 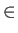 [1,nspecs] in ascending order with
[1,nspecs] in ascending order with 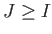 . Note that n_I is expected only when
. Note that n_I is expected only when 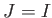 , with n_I specifying the number of atoms for each species I 1. The value for n_1 (= n_A) is inferred from the knowledge of the total number of atoms (NATOMS). For example, a ternary system A
, with n_I specifying the number of atoms for each species I 1. The value for n_1 (= n_A) is inferred from the knowledge of the total number of atoms (NATOMS). For example, a ternary system A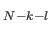 B
B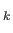 C
C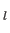 (N = NATOMS) should be specified in six consecutive lines:
(N = NATOMS) should be specified in six consecutive lines:
| MGUPTA |
3 |
A_AA |
p_AA |
q_AA |
xi_AA |
r0_AA |
| MGUPTA |
|
A_AB |
p_AB |
q_AB |
xi_AB |
r0_AB |
| MGUPTA |
|
A_AC |
p_AC |
q_AC |
xi_AC |
r0_AC |
| MGUPTA |
k |
A_BB |
p_BB |
q_BB |
xi_BB |
r0_BB |
| MGUPTA |
|
A_BC |
p_BC |
q_BC |
xi_BC |
r0_BC |
| MGUPTA |
k |
A_CC |
p_CC |
q_CC |
xi_CC |
r0_CC |
- MIE_FIELD filename Rc Bx By Bz: specifies a fixed substrate from superposition of Mie-type central force fields, each defined by
The parameters , , , and coordinates of each Mie site must be provided in the file filename. The expected file format is:
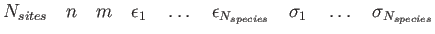
where and are given for each species (e.g. for nanoalloys). Mie field(s) can be smoothly truncated using the optional cut-off parameter Rc, which will trigger the usual Stoddard and Ford procedure:
Periodic boundary conditions with the nearest image convention can be imposed on the field sites using the optional parameters Bx, By and Bz, which specify the periodic box dimensions.
- MIN_ZERO_SEP separation attempts: when used in combination with the FEBH keyword, this will tighten the rms force convergence thresholds until
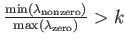 where
where 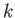 is the value given by separation. If attempts is specified, the GMIN run will
terminate if the separation is not achieved within the specified number of attempts. This is usually indicative of a problem with the potential or eigenvalue calculation.
is the value given by separation. If attempts is specified, the GMIN run will
terminate if the separation is not achieved within the specified number of attempts. This is usually indicative of a problem with the potential or eigenvalue calculation.
- MLJ nspecs eps_11 sig_11: specifies a multicomponent Lennard-Jones potential for nspecs distinct species. If nspecs = 1 then this single line is sufficient, and it ought to yield the same results as the standard Lennard-Jones. Extension to nspecs 1 is analogous to the MGUPTA keyword, and it differs from the input format expected by the GLJ keyword. Note that, although GLJ and MLJ link to different implementations of the general multicomponent Lennard-Jones, they ought to yield the same results.
- MSC nspecs nn11 mm11 sig11 sceps11 scc1: specifies a multicomponent Sutton-Chen potential for nspecs distinct metallic species. If nspecs = 1 then this single line is sufficient, and it can be used in conjunction with keywords PERIODIC and CUTOFF. The parameters nn11, mm11, sig11, sceps11 and scc11 have the same meaning as for the keyword SC. If a cutoff has been specified, then the potential will be smoothly truncated by a generalised Stoddard and Ford procedure, which ensures the energy and its first derivatives remain continuous. If nspecs 1, then
more subsequent lines of the form MSC (ntypeI) nnIJ mmIJ sigIJ scepsIJ (sccI) must be supplied for all the remaining I,J [1,nspecs] in ascending order with . The parameters ntypeI and sccI are expected only when , with ntypeI specifying the number of atoms for each species I
 1. The value for ntype1 (= ntypeA) is inferred from the knowledge of the total number of atoms (NATOMS). For example, a ternary system ABC (N = NATOMS) should be specified in six consecutive lines:
1. The value for ntype1 (= ntypeA) is inferred from the knowledge of the total number of atoms (NATOMS). For example, a ternary system ABC (N = NATOMS) should be specified in six consecutive lines:
| MSC |
3 |
nnAA |
mmAA |
sigAA |
scepsAA |
sccA |
| MSC |
|
nnAB |
mmAB |
sigAB |
scepsAB |
|
| MSC |
|
nnAC |
mmAC |
sigAC |
scepsAC |
|
| MSC |
k |
nnBB |
mmBB |
sigBB |
scepsBB |
sccB |
| MSC |
|
nnBC |
mmBC |
sigBC |
scepsBC |
|
| MSC |
l |
nnCC |
mmCC |
sigCC |
scepsCC |
sccC |
- MSORIG : specifies a particular tight-binding potential for silicon.
- MSTRANS : specifies an alternative tight-binding potential for silicon.
- MULLERBROWN : specifies the 2D Muller-Brown potential.
- MULTIPERM : instead of basin-hopping, systematically span the permutation space of the multiset containing particle labels. Uses a more general algorithm than ENPERMS .
- MULTIPLICITY xmul: specifies the multiplicity of the electronic state in DFTB
calculations.
- MWPOT: calls a Stillinger-Weber type potential with parameters appropriate for water[23].
- NATB: specifies the sodium tight-binding potential of Calvo and Spiegelmann.
This potential can be guided by also specifying GUPTA 21 in the data file.
- NCAP: calls the capsid model [24], consisting of pentagonal building blocks.
- NEON: introduces a diatomics-in-molecules calculation for
a neutral, cationic or electronically excited neon cluster. See also
GROUND, PLUS, TWOPLUS and STAR.
- NEWJUMP prob: for (serial)
`parallel' runs specifies a jump probability between runs
(parallel tempering) of prob.
See the BHPT keyword for a better alternative.
- NEWRESTART nrelax nhs MD newrestemp: reseed runs if the energy does not decrease within nrelax steps.
nhs is the number of hard sphere moves used to produce the new starting configuration.
If nhs=0 (the default) then the geometry is changed by reseeding. If MD is present, then a short AMBER or CHARMM MD run is performed at temperature newrestemp (specified in K, default 1000K) to generate the new configuration.
If the `F' argument appears for AVOID then
NEWRESTART will not reseed.
- NEWTSALLIS q: specifies that steps are accepted/rejected using Tsallis statistics with the
given value of q, rather than the usual Boltzmann condition. This version is slightly different from "TSALLIS" keyword.
- NOCHIRALCHECKS: disables checks for inversion of CA atoms and chiral side-chains for ILE and THR.
- NOCISTRANSCHECKS: disables checks for isomerisation of the peptide bond. This is equivalent to using the CISTRANS keyword.
- NOCISTRANS minomega: set on by default with a threshold minomega of 150 degrees.
If an amide bond is deformed to a angle below the specified threshold, the structure is discarded.
i.e. with
 minomega. minomega defaults to 150 degrees.
For proline every
minomega. minomega defaults to 150 degrees.
For proline every 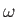 is allowed. To enable cis-trans isomerisation, the CISTRANS keyword should be used.
is allowed. To enable cis-trans isomerisation, the CISTRANS keyword should be used.
- NOCISTRANSDNA minomega: should be specified when working with DNA in AMBER to ensure the correct bonds are
checked. As above, the deformation threshold minomega can be set. It is defaulted to 150 degrees.
- NOCISTRANSRNA minomega: should be specified when working with RNA in AMBER to ensure the correct bonds are
checked. As above, the deformation threshold minomega can be set. It is defaulted to 150 degrees.
- NOFREEZE: don't freeze the core atoms when doing the initial geometry optimisations in
a run where SEED is specified.
- NOINVERSION: turns off inversion of structures for distance
metric in minpermdist.
- NOPHIPSI: used with the CHARMM keyword to specify twisting of
sidechain dihedrals only.
- NORESET: by default the configuration point is set to that of the
quench minimum in the Markov chain during a basin-hopping simulation. This
keyword turns off the resetting so that the geometry varies continuously.
- NOTE : the rest of the line is ignored.
- NPAH n: calls a finite system of polycyclic aromatic hydrocarbons (PAH) interacting via
Williams' potential [25]. The PAH ID n defines the PAH molecule: 1 for coronene, 2 for
pyrene, 3 for benzene.
- NRELAXRIGID MinR MinA: Minimizations will use rigid body coordinates for MinR steps and then atomistic coordinates for MinA steps. This is to be used with RIGIDINIT keyword.
- NTIP n: calls a member of TIP family of potentials for water molecules within a rigid-body
framework.
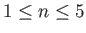 .
. 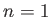 : TIPS water;
: TIPS water; 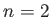 : TIPS2 water;
: TIPS2 water; 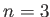 : TIP3P water;
: TIP3P water;
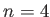 : TIP4P water; and
: TIP4P water; and 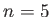 : TIP5P water.
: TIP5P water.
- ODIHE : order parameter--requires documentation.
- OEINT : interaction energy between 2 peptides will be used as an order parameter--requires
further documentation.
- OHCELL : allow point group operations for a cubic
supercell in subroutine minpermdist.f90 permutational distance minimisation.
- ORGYR : radius of gyration will be calculated as an order parameter--requires
further documentation.
- OSASA : order parameter--requires documentation.
- P46: specifies a 46-bead three-colour model polypeptide.
See also the BLN keyword, which implements this potential in a more
general way and uses unit bond lengths.
- PACHECO: specifies the intermolecular Pacheco-Ramelho potential for C.
The Axilrod-Teller contribution, specified with the AXTELL keyword, is included
when the RMS force falls below the value entered with GUIDECUT.
- PAH: specifies a polycyclic aromatic hydrocarbon potential.
- PAHA n: calls a finite system of polycyclic aromatic hydrocarbons (PAH) interacting
via a general anisotropic potential developed from first principles [26]. The PAH ID
n defines the PAH molecule: 1 for benzene, 2 for naphthalene, 3 for anthracene, and 4 for
pyrene.
- PAIRDIST pair1a pair1b pair2a pair2b...: enables tracking of the distances between pairs of atoms during a GMIN run. Atom pairs may
be specified in the keyword definition as shown, or in the file pairdist with a pair of atoms on each line. The pair distances are
calculated after each quench and printed in pairdists.
- PAP npatch alpha s cosdel epsilon: specifies a patch anti-patch potential. Each body consists of a Lennard-Jones core, range parameter alpha, with npatch patches and npatch anti-patches. The patch anti-patch attraction has a range specified by s, a width specified by cosdel and a strength specified by epsilon. If npatch is set to zero, site information will be read in from the file papsites.xyz.
- PARALLEL npar GMIN: npar is the number of parallel runs within GMIN.
If the word "GMIN" is present as the second argument, the calculation will proceed until the
same lowest energy is recorded in each trajectory.
- PBGLUE: specifies a glue potential for lead.
- PERCOLATE dist comp cutoff: specifies that particles are prevented from evaporating using a system based on maintaining a percolating graph of particles, rather than using a container, with dist as the maximum distance at which particles are considered to be connected. An optional harmonic compression potential can be specified with force constant comp, to be turned off below RMS force cutoff. Currently, the step size is limited to dist squared.
- PERIODIC boxlx boxly boxlz: specifies periodic boundary conditions for
potentials which understand such a directive (such as tight-binding silicon). The three
double precision variables are the box lengths. If only one box length is given the
others are set to the same value to give a cube.
- PERMDIST x: minimise distances between
the coordinates in files coords and the
fixed coordinates in file finish with respect to permutational isomerisation.
Requires the auxiliary file perm.allow to specify permutable atoms, otherwise
all atoms are assumed to be permutable. The absence of a perm.allow
file is considered a mistake for CHARMM runs.
The parameter x is the distance tolerance for assigning atoms to orbits
in the myorient standard orientation routine, default value 0.001.
If x is too small it is possible for permutational isomers to be missed,
however, if x is too large then runs with the AVOID keyword can
slow down by an order of magnitude.
The first line of the perm.allow file must contain an integer
that specifies the number of primary groups of interchangeable atoms.
The groups then follow, each one introduced by a line with two integers 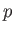 and
and 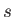 that specify the number of permutable atoms in the primary group and the number of other sets
of permutable atoms associated with the primary set.
may be zero.
Each secondary set of permutable atoms has members.
The following line contains the indices of the permutable atoms
in the primary set and then
the indices of the atoms in each of the secondary sets, one set at
a time.
that specify the number of permutable atoms in the primary group and the number of other sets
of permutable atoms associated with the primary set.
may be zero.
Each secondary set of permutable atoms has members.
The following line contains the indices of the permutable atoms
in the primary set and then
the indices of the atoms in each of the secondary sets, one set at
a time.
For the phenylalanine example illustrated we must allow three other pairs of
atoms to exchange if we swap 7 and 8. Hence a suitable perm.allow entry is
1
2 3
7 8 5 6 1 2 3 4
Here and  : if we exchange 7 and 8 then we must also exchange 5 and 6,
1 and 2, and 3 and 4. There are two atoms in each of the three secondary sets,
since we have specified 7 and 8 as the two primary atoms.
: if we exchange 7 and 8 then we must also exchange 5 and 6,
1 and 2, and 3 and 4. There are two atoms in each of the three secondary sets,
since we have specified 7 and 8 as the two primary atoms.
Here is an example perm.allow file for a water trimer using
the flexible QTIP4PF potential, where the energy is invariant to permutations
of water molecules and to exchanges of hydrogens in the same molecule. However,
hydrogens cannot exchange between different oxygens:
4
3 2
1 4 7 2 3 5 6 8 9
2 0
2 3
2 0
5 6
2 0
8 9
The first group of three oxygens has two atoms that must move with each oxygen,
i.e. atoms 2 and 3 for oxygen 1, etc. Hydrogen permutations for each oxygen are
allowed by the three following groups. This scheme allows atoms to appear in more
than one group. There must be a group containing each complete set of permutations
in order for permutation-inversion isomers to be recognised. The format
is compatible with an older scheme, where only pair swaps were allowed for
associated atoms, but now allows for more general permutations.
Scripts to generate allowed permutations automatically for CHARMM and AMBER are available from
the group web site. It is essential to use symmetrised versions of the corresponding
force fields!
- PERMINVOPT x: minimise the distance between the geometries in
coords and finish with respect to permutations, as well
as overall translation, rotation, and inversion. See
PERMOPT for distance optimisation without the inversion.
To specify specific groups of
permutable atoms a perm.allow file is needed, unless all atoms (or rigid bodies)
are considered permutable.
For rigid bodies the file rbsymops can be used to specify symmetry
operations of each molecule, and the distance will then also be
minimised with respect to internal symmetry operations.
To minimise the Euclidean distance between two configurations with no permutations
a perm.allow file with the single entry `0' can be used.
The configurations are then aligned as if they were rigid bodies decorated
with fixed sites.
The parameter x is the distance tolerance for assigning atoms to orbits
in the myorient standard orientation routine, default value 0.001.
If x is too small it is possible for permutational isomers to be missed,
however, if x is too large then runs with the AVOID keyword can
slow down by an order of magnitude.
- PERMOPT x: minimise the distance between the geometries in
coords and finish with respect to permutations, as well
as overall translation and rotation, but not the inversion. To specify specific groups of
permutable atoms a perm.allow file is needed, unless all atoms (or rigid bodies)
are considered permutable.
For rigid bodies the file rbsymops can be used to specify symmetry
operations of each molecule, and the distance will then also be
minimised with respect to internal symmetry operations.
To minimise the Euclidean distance between two configurations with no permutations
a perm.allow file with the single entry `0' can be used.
The configurations are then aligned as if they were rigid bodies decorated
with fixed sites.
The parameter x is the distance tolerance for assigning atoms to orbits
in the myorient standard orientation routine, default value 0.001.
If x is too small it is possible for permutational isomers to be missed,
however, if x is too large then runs with the AVOID keyword can
slow down by an order of magnitude.
- PLUS: when combined with keywords NEON or ARGON
uses a diatomics-in-molecules potential for the singly charged cation.
- POWER ipow: ipow is the initial power for shifts in the old line minimisation routine
for conjugate gradient. LBFGS minimisation should be used instead.
- PROJI: turns on projection operator to enforce
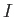 point group symmetry
in mylbfgs.f. The geometry is projected after every proposed step.
point group symmetry
in mylbfgs.f. The geometry is projected after every proposed step.
- PROJIH: turns on projection operator to enforce
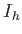 point group symmetry
in mylbfgs.f. The geometry is projected after every proposed step.
point group symmetry
in mylbfgs.f. The geometry is projected after every proposed step.
- PRTFRQ n: prints the energy every n steps; default is every step.
Should now work for basin-hopping, basin-sampling and parallel tempering runs.
- PRINT_PTGRP tol1 tol2 tol3: Print point-group classification and order for each structure saved in the file lowest. The three tolerances are optional: tol1 is a dimensionless tolerance for diagnosing degeneracies in (normalised!) principal moments of inertia, tol2 is the distance tolerance used for detecting point-group symmetry operations, and tol3 is a dimensionless tolerance used for testing whether a generated rotation matrix (representing a symmetry operation) is new. The default values are
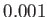 ,
, 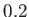 and
and 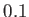 , respectively.
, respectively.
- PTMC histmin histmax ptemin ptemax pttmin pttmax exchprob nequil ptsteps nenrper hbins:
requests a standard parallel tempering MC run.
This keyword also specifies the energy range for the histogram of quench energies,
histmin to histmax,
the energy range for the histogram of instantaneous configurations, ptemin to ptemax,
the temperature range (pttmin and pttmax),
the probability of attempting an exchange exchprob, the
number of equilibration steps, nequil,
the number of parallel tempering MC steps without quenching, ptsteps,
the number of bins for the histogram of instantaneous potential energy, nenrper, and
the number of bins for the histogram of quench energies, hbins.
Should be used together with the MPI keyword.
(This option is only available if the source is compiled with an MPI enabled.)
- PULL a1 a2 f: apply a static force to the potential, equivalent to adding
the term
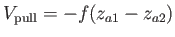 . Here
. Here 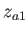 and
and 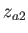 are the
coordinates for atoms
are the
coordinates for atoms 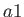 and
and 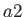 , and
, and 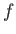 specifies the force.
This potential is designed to simulate a pulling experiment with static force where
a molecule is pulled along the axis from atoms and .
specifies the force.
This potential is designed to simulate a pulling experiment with static force where
a molecule is pulled along the axis from atoms and .
- PY sig0 eps0 [cutoff XYZ xbox ybox zbox]: specifies a rigid-body multisite Paramonov-Yaliraki[27] potential with parameters and
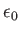 . Optional arguments are
a cutoff distance in absolute units, a specification of which directions have periodic boundary
conditions, and the size of the periodic box in units of the cutoff. The parameters for and setup
of the rigid body, even if it is just one site, must be specified in a file pysites.xyz.
. Optional arguments are
a cutoff distance in absolute units, a specification of which directions have periodic boundary
conditions, and the size of the periodic box in units of the cutoff. The parameters for and setup
of the rigid body, even if it is just one site, must be specified in a file pysites.xyz.
There are two formats for pysites.xyz: identical particles and n-ary mixtures. For identical
particles, the format is:
[number of sites in molecule]
[blank line]
site [x position] [y] [z] shapes [rep coeff] [att coeff] [a11] [a12] [a13] [a21] [a22] a[23] orient [p_x] [p_y] [p_z]
...
site [x position] [y] [z] shapes [rep coeff] [att coeff] [a11] [a12] [a13] [a21] [a22] a[23] orient [p_x] [p_y] [p_z]
For an n-ary mixture,
0 [the number zero]
[arity]
[blank line]
[number fraction of this type of molecule]
[number of sites in molecule]
[normal site lines]
...
[blank line]
[second number fraction]
[number of sites in second type]
[normal site lines]
...
For example, for a single site molecule,
1
site 0.0 0.0 0.0 shapes 1.0 1.0 0.5 0.5 0.2 0.5 0.5 0.2 orient 0.0 0.0 0.0
Or, for example, for two-to-one mixture of single sites and pairs,
0
2
0.66
1
site 0.0 0.0 0.0 shapes 1.0 1.0 0.5 0.5 0.2 0.5 0.5 0.2 orient 0.0 0.0 0.0
0.34
2
site 0.0 0.5 0.0 shapes 1.0 1.0 0.5 0.5 0.2 0.5 0.5 0.2 orient 0.0 0.2 0.0
site 0.0 -0.5 0.0 shapes 1.0 1.0 0.5 0.5 0.2 0.5 0.5 0.2 orient 0.0 -0.2 0.0
- QALCS qalcsmode cutoff: perform Quench-Assisted Local Combinatorial Search to locate a biminimum for a multicomponent system. (More general than HOMOREF, which works only for binary systems.) The search comprises a sequence of quench-assisted atom swaps, where each step involves a sweep through the local neighbourhood structure induced by the ``interchange'' distance metric. There are many ways of doing this: qalcsmode=0 specifies a slow steepest-descent-like search in permutation space; whereas modes 1 through 5 specify alternative schemes that are more efficient for larger systems. Modes 4 and 5 admit an additional (optional) integer parameter cutoff, which specifies that only the first cutoff entries in the sorted local neighbourhood are to be considered. The sorting is done by approximate swap gain.
- QALCS_SURF: specifies that QALCS should include surface vacancies as a separate species, akin to dynamic-lattice-search-type methods but relies on a different construction of surface vacancies. The scheme constitutes a deterministic way of refining the surface of a cluster within the QALCS framework, and it can be used with or without the
QALCS keyword (for single- or multi-component clusters). Note that during the surface optimisation all particles are treated as being the same.
- QMAX cgmax: cgmax is the tolerance for the
RMS force in the final set of quenches that are used to produce
the output for file lowest. The default is
cgmax
 , but the appropriate value depends upon the system in question.
TIGHTCONV can be used instead.
, but the appropriate value depends upon the system in question.
TIGHTCONV can be used instead.
- QUAD: requires documentation.
- QUCENTRE: sets the centre of coordinates to the origin (0,0,0) before each MC step is taken (so after each quench), but not during the minimisation itself unlike CENTRE.
- RADIUS radius: sets the radius of the container that prevents particles
evaporating during quenches. If unset the program calculates an appropriate value
based upon the volume per particle for close-packed material and the known pair
equilibrium distance for the given potential. The formula employed is
where is the number of atoms and  is the pair equilibrium
separation.[28] The `1' in this formula is to allow some extra space for
more open structures.
is the pair equilibrium
separation.[28] The `1' in this formula is to allow some extra space for
more open structures.
- RANDOMSEED: specifies that the random number generator should be seeded with system time after each quench, allowing simple parallel use. Currently functional only for the CHARMM and AMBER potentials.
- RANSEED i: integer seed for the random number generator. The number actually used is mod(i,10000)+1.
- RANDPERM: swap coordinates of randomly-picked unlike atom pairs in a binary system. The total number of pair swaps is randomly chosen from the interval
![$ [0.5*N, N]$](img167.png) , where
, where
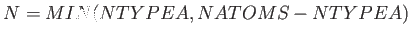 , and then rounded up to the nearest integer. This can be used as a ``random permutation''-type move (and in conjunction with a random cartesian displacement).
, and then rounded up to the nearest integer. This can be used as a ``random permutation''-type move (and in conjunction with a random cartesian displacement).
- RANDMULTIPERM n: randomly permute atomic labels every n basin-hopping steps. Intended for use in conjunction with keyword QALCS.
- RATIO stepratio tempratio: adjusts stepsize and temperature independently.
stepratio is the target fraction of steps that move into a different well. Identity of
structures is determined by structural alignment; as such, the PERMDIST keyword and
appropriate auxiliary files are required. tempratio is the target acceptance ratio for
those steps that move into new basins. If a negative number is supplied for either, the
ratios is printed, but the corresponding parameter is not adjusted.
- RBSYM: specifies that internal symmetry operations permuting equivalent sites for
rigid-body building blocks exist. The operations are read in from a file rbsymops, which must
exist in the working directory. The first line of the file is an integer that specifies the number
of operations generated by proper axes of rotation including the identity operation; the operations
are then read in one at a time in a representation consisting of a unit vector, defining the
axis in the reference frame, and an angle in degrees, describing the magnitude of rotation about
that axis. The first operation has to correspond to the identity operation.
This keyword has to be combined with PERMDIST so that structures are aligned and
a distance metric considered based on permutations of identical rigid
bodies and any internal symmetry operation of each rigid body that is a symmetry
of the potential energy function.
- RELAXFINALQUENCH: This means final quench is done in atom coordinates. Useful if you want to compare final energies. This is to be used with RIGIDINIT keyword.
- RESERVOIR n: for use in combination with PTMC. A reservoir of local minima
is used for the lowest temperature replicas up to cpu number n. The default is
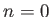 , which means that only the lowest replica used the reservoir. Visit statistics are
dumped for replicas that use the reservoir, but exchanges only occur when the lowest
non-reservoir replica inherits a configuration. This keyword requires files
min.data and points.min in the current working directory in PATHSAMPLE
format. See also EXEQ.
, which means that only the lowest replica used the reservoir. Visit statistics are
dumped for replicas that use the reservoir, but exchanges only occur when the lowest
non-reservoir replica inherits a configuration. This keyword requires files
min.data and points.min in the current working directory in PATHSAMPLE
format. See also EXEQ.
- RESIZE resize: all the coordinates are multiplied by resize after
they have been read in, before any other operations are performed. This command is useful
for scaling results obtained with one potential for a system with a different pair
equilibrium distance.
- RESTART nrelax nhs: reseed runs if a step in not accepted
within twice nrelax steps.
nhs is the number of hard sphere moves used to produce the new starting configuration.
If nhs=0 (the default) then the geometry is changed by reseeding.
- RESTORE dumpfile intEdumpfile: restore a previous GMIN run from a dumpfile.
The number of basin-hopping steps performed will be the difference between the number
requested for the run that produced the dumpfile, minus the number that were completed
at the point the dumpfile was created. This option is not available before version 2.3.
If you are using the A9INTE keyword, you can specify the interaction enthalpy
dump file to restore from as a second arguement.
- RGCL2: specifies a DIM rare gas-Cl potential.
- RIGIDINIT: This will turn on the local rigid body framework. This keyword needs two additional input files: coordsinirigid and rbodyconfig. coordsinirigid should contain the coordinates of all the atoms whether they are part of local rigid bodies or not. During initialization, GMIN will select appropriate coordinates and throw away irrelevant ones. The format is:
x1 y1 z1
x2 y2 z2
ldots
ldots
xn yn zn
The file rbodyconfig defines the rigid bodies, with the following format:
GROUP NoAtomsInGroup
Atom 1
Atom 2
ldots
ldots
Atom N
The keywords RELAXFINALQUENCH and NRELAXRIGID can be used in conjuction with RIGIDINIT.
- RINGROTSCALE factor: when applying cartesian moves with CHARMM, amino acid rings are moved as rigid units. Setting factor (default 0.0) between 0.0 and 1.0 will apply a random rotation to these rings during step taking. The suggested value is 0.1 to prevent the regular formation of high energy structures.
- RKMIN: specifies a Runga-Kutta minimisation scheme.
Very inefficient.
- RMS rmslimit rmstol rmssave lca best: used with CHARMM keyword to
specify that the RMSD compared to a reference geometry is calculated. The reference geometry must
be given in xyz-format in an additional file compare. rmssave is an integer
that specifies the number of lowest energy geometries and RMSD
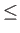 rmslimit
to save. Geometries are only saved if their RMSD's are more than rmstol
different. The flag lca controls whether the all-atom RMSD (lca=0) or the
rmslimit
to save. Geometries are only saved if their RMSD's are more than rmstol
different. The flag lca controls whether the all-atom RMSD (lca=0) or the
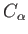 -RMSD
(lca=1) is calculated. The flag best determines which structure is compared to the reference
after each quench. best=0 implies the current quench minimum and best=1 implies the current best (lowest energy) minimum. If TRACKDATA is also specified, the RMSD calculated after each quench is produced in the file `rmsd' in gnuplot readable format.
-RMSD
(lca=1) is calculated. The flag best determines which structure is compared to the reference
after each quench. best=0 implies the current quench minimum and best=1 implies the current best (lowest energy) minimum. If TRACKDATA is also specified, the RMSD calculated after each quench is produced in the file `rmsd' in gnuplot readable format.
- ROTAMER maxchange pselect occuw (centre cutoff): Used with AMBER9 only. Specifies that rotamer moves should be taken. Every step, up to maxchange rotamers may be selected with a probability pselect. occuw determines the minimum % occupation a rotamer must have to be selected from the library[29] when making a change. For example, occuw
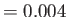 restricts possible rotamer choice to those with a greater than 0.4% occupation. If you want to focus rotamer changes around a ligand/binding pocket, the optional centre and cutoff arguements may be used. centre specifies the residue of interest (for example a ligand), and cutoff the limiting distance from this centre that rotamers may be changed. The selection probability decreases linearlly from the centre residue. To use these moves, you need three files found in the SCRIPTS directory: PdbRotamerSearch, penultimate.lib and rotamermove.csh in your working directory for each run.
restricts possible rotamer choice to those with a greater than 0.4% occupation. If you want to focus rotamer changes around a ligand/binding pocket, the optional centre and cutoff arguements may be used. centre specifies the residue of interest (for example a ligand), and cutoff the limiting distance from this centre that rotamers may be changed. The selection probability decreases linearlly from the centre residue. To use these moves, you need three files found in the SCRIPTS directory: PdbRotamerSearch, penultimate.lib and rotamermove.csh in your working directory for each run.
- ROTATERIGID freq factor: for use with the generalised rigid body framework,
RIGIDINIT. Randomly rotates each rigid body in the system by a factor of
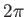 multiplied by factor every freq steps.
multiplied by factor every freq steps.
- SANDBOX: Specifies that the sandbox potential should be used. This keyword requires an auxiliary input file rbdata and produces an auxiliary output visualization file sandout.xyz. See the Sandbox section of this documentation for information on producing rbdata files and for examples.
- SAVE nsave: nsave is an integer that specifies the number of lowest
energy geometries to save and summarise in the file lowest.
Arrays are now dynamically allocated, so any positive integer can be specified. Note that if nsave is large, minima that are not in the Markov chain might also be written out which might be high energy non-physical conformations.
- SAVEMULTIMINONLY: specifies that only multiminima are to be considered for the final dump to lowest.
- SAVEINTE nsaveinte: nsaveinte is an integer that specifies the number of lowest
interaction enthalpy geometries to save and summarise in the file intelowest. See A9INTE.
- SEMIGRAND_MU mu_1 ...mu_M: specifies the semi-grand chemical potentials, relative to the first species, for use in conjunction with the keyword LFLIPS. The number of values ought to match the number of different species minus one (i.e. M=NSPECIES-1).
- SETCENTRE x y z: Sets the centre of mass/coordinates (before the initial quench) to (x,y,z). For example, SETCENTRE 0.0 0.0 0.0
would translate the centre of mass to the origin.
- SETCHIRAL: For use with AMBER9, NAB and AMBER12 keywords. Specifies that the expected chirality of each centre should be
maintained based on that in the initial quenched minimum, rather than the standard chiralities found in most proteins/nucleotides.
WARNING: Presently for AMBER12, without this keyword being included, no chirality checking is done at all!
- SC nn mm sig sceps scc: specifies a Sutton-Chen potential[30] with
parameters
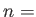 nn,
nn, 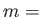 mm,
mm, 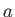 =sig,
=sig, 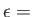 sceps and
sceps and
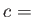 scc.
scc.
- SEED nsstop: if the SEED keyword appears then the program
looks for a file seed containing coordinates, which are used to `seed' the new run.
The number of coordinates given in this file should be no more than one less than the number
given in coords. The specified coordinates are frozen from the first step until
step nsstop.
- SHIFTCUT: specifies a shifted-truncated potential for bulk binary Lennard-Jones.
- SLOPPYCONV bgmax: specifies a basin-hopping run (as opposed to standard MC
on the untransformed surface). bgmax is the convergence criterion
for the RMS force in the basin-hopping
quenches. If this criterion is too strict then the run time will be greatly increased.
If it is too sloppy then the performance of the algorithm is impaired. Different values
are needed for different potentials. BASIN can be used instead.
- SORT: for pairwise potentials the atoms can be sorted from most to least
strongly bound. The SORT keyword enables this sorting for the coordinates printed
in file lowest. This can be useful for seeding subsequent runs by removing the
most weakly bound atoms. This sort is not set by default and is meaningless if the
pair energies are not computed.
- SPECLABELS L1 L2 ...LM : specifies the labels to be used in file lowest for each atomic species. Intended for use with a potential invoked by keywords MLJ, MGUPTA and MSC. Note that each label is interpreted as a string of two characters, and the calculation will stop if the supplied number of labels (M) does not match the species count.
- STAR: specifies an excited state calculation for Ar
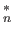 or Ne for
a diatomics-in-molecules potential when used with NEON or ARGON.
or Ne for
a diatomics-in-molecules potential when used with NEON or ARGON.
- STEP step astep ostep block: specifies the maximum step sizes. step is
for the maximum change of any Cartesian coordinate and astep specifies a tolerance
on the binding energy of individual atoms (if available, i.e. for Morse and LJ) below
which an angular step is taken for that atom. See the following section for more details.
ostep is the maximum displacement of an axis-angle coordinate for a rigid body system
and block (an integer) is the block size for which separate translational and orientational
displacements will be made for rigid bodies. Omitting block or using a value of zero results in
translational and orientational steps being taken simultaneously
for rigid bodies. The default values for step,
astep and ostep are all 0.3 and the default value of nblock is zero.
- STEEREDMIN smink sminkinc smindiststart smindistfinish sminatoma sminatomb: specified steered
minimisation should be performed (must be used with AMBER9). For a protein/ligand system, this adds a translation
to the MC move. The vector between the centre of coordinates of groups A and B (as defined in the file movableatoms)
is calculated and set to smindiststart. During the following minimisation, a restoring force is applied to
the ligand. The harmonic force constant is initially zero, and rises by sminkinc every LBFGS step up to a
maximum of smink. The force is applied until the A->B distance is less than smindistfinish.
- STEPS mcsteps tfac: determines the length of the
basin-hopping run through the integer mcsteps and the annealing protocol through
the real variable tfac. The temperature is multiplied by tfac
after every step in each run.
- STICKY nrbsites, sigma: specifies a `sticky patch' potential with nrbsites
sites in the rigid body reference and a value of sigma for the parameter.
- STOCK mu lambda: specifies a Stockmeyer potential with parameters
and , respectively.
- STOCKAA muD E: calls a finite system of Stockmayer particles,
where is the dipole moment strength and the electric field of stength can be optionally present. The field, if
present, is along the space-fixed z-direction.
- STRAND: specifies a system of
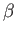 strands coded using the rigid body formalism.
strands coded using the rigid body formalism.
- SUPPRESS: suppresses the majority of the GMIN output.
Think of it as the opposite of DEBUG.
- SW: specifies the Stillinger-Weber Si potential.
- SYMMETRISE int tol1 tol2 tol3 tol4 tol5 qmax mdiff d: specifies that the symmetrisation
routine should be called every int steps. The five tol parameters are tolerances
for various parts of the routine:
tol1 is used in ptgrp.f in defining orbits;
tol2 is the distance tolerance used in ptgrp.f to define point group symmetry operations;
tol3 is the maximum relative difference in principal moments of inertia used to
diagnose point groups with degenerate irreducible representations in ptgrp.f;
tol4 is the distance cutoff used to determine if a symmetry element has been lost in symmetry.f.
Since we are dealing with approximate symmetries, this parameter may be larger than tol2.
It is compared to the largest atomic displacement divided by the corresponding radius
for the closest permutation.
tol4 is also used to test whether atoms lie on a given symmetry element, and in testing
whether orbits generated from `floaters' are actually contained in the core.
tol5 is generally to check for atom clashes in symmetry.f, including analysis of
missing sites in orbits, as well as overlap between orbits generated from `floaters' and
previous core or new orbit sites.
qmax is the maximum number of quenches allowed for each call to symmetry.f.
mdiff is used to test whether a generated symmetry operation is new. If any of the nine
components of the corresponding
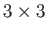 matrix differs by more than mdiff from an
existing matrix then the operations are considered to be different.
d is the exponential factor used in constructing a centre of mass that is biased towards
core atoms. The contribution of each atom is weighted by
matrix differs by more than mdiff from an
existing matrix then the operations are considered to be different.
d is the exponential factor used in constructing a centre of mass that is biased towards
core atoms. The contribution of each atom is weighted by
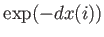 , where is the
centre of mass distance of atom on the previous cycle.
, where is the
centre of mass distance of atom on the previous cycle.
- TABOO nlist: specifies a taboo list of the nlist lowest minima should be maintained.
- TARGET target1 target2
 : specifies any number of target energies.
The current run stops in an orderly
fashion if the current quench energy is within econv of any target (see EDIFF).
: specifies any number of target energies.
The current run stops in an orderly
fashion if the current quench energy is within econv of any target (see EDIFF).
- TEMPERATURE temp: defines the temperature, temp, at which the
MC runs are conducted. Different values can be specified for serial `parallel' runs if
PARALLEL is set.
For true parallel basin-hopping use the BHPT keyword and omit TEMPERATURE.
- TETHER hdistconstraint hwindows ExtrapolationPercent lnHarmFreq: requests a calculation of the vibrational density of
states for a given minimum. hdistconstraint is the minimised average radius of the basin of attraction to which the minimum
belongs, hwindows is the number of potential energy windows into which a WL simulation is split. ExtrapolationPercent is the percentage of the whole potential energy spectrum, for which the density of states is estimated from
the harmonic approximation and not sampled. lnHarmFreq the log product of positive Hessian
eigenvalues.
- THOMSON q: specify the Thomson problem for unit charges on a sphere.
If q is present it is taken to be the charge on one particle, which can
therefore be different from all the other unit charges and is read as a real number.
- TIGHTCONV cgmax: cgmax is the tolerance for the
RMS force in the final set of quenches that are used to produce
the output for file lowest. The default is
cgmax, but the appropriate values depend upon the system in question.
QMAX can be used instead.
- TIP n: specifies a TIPnP intermolecular potential for rigid body water molecules.
 .
.
- TOLBRENT tolb: parameter for DBRENT minimisation.
Inefficient compared to LBFGS.
- TOMEGA: used with the CHARMM keyword to specify that peptide bonds will be twisted along with all other dihedrals.
- TOSI app amm apm rho: specifies the Tosi-Fumi potential[31]
with parameters
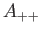 ,
, 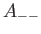 ,
, 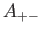 and .
and .
- TRACKDATA: produces `energy.dat' and `markov.dat' containing the quench number and
associated energy and markov energy in two columns and `best.dat', containing the current quench number and the current lowest
total energy. If RMS is also specified, a file called `rmsd.dat' is produced containing the RMSD from a reference structure.
See RMS for more information. This allows plotting with gnuplot to monitor convergence of multiple runs.
If A9INTE is also specified, two additional output files are produced, `intE.dat' containing the quench number and associated interaction
enthalpy, and `bestintE.dat' containing the quench number and current lowest interaction enthalpy. This keyword does not yet function for MPI runs.
- TRANSLATERIGID freq maxdist: for use with the generalised rigid
body framework, RIGIDINIT. Randomly translates each rigid body in the
system by a distance up to maxdist every freq steps.
- TSALLIS q: specifies that steps are accepted/rejected using Tsallis statistics with the
given value of q, rather than the usual Boltzmann condition.
- TTM3: specifies Xantheas' TTM3-F water potential, coded
by Jeremy Richardson.
The atom order must be O1, O2,..., H1a, H1b, H2a, H2b,....
The energy is in kcal/mol and the distance unit is Å.
Hydrogens are NOT permutable between different oxygens.
Note that this potential is prone to cold fusion--setting the COLDFUSION
parameter explicitly may be necessary.
- TWOPLUS: when combined with keywords NEON or ARGON
uses a diatomics-in-molecules potential for the doubly charged cation.
- UACHIRAL: MUST be included when using ff03ua, the AMBER united atom forcefield unless you have disabled the checks for inverted chiral carbons
with NOCHIRALCHECKS. UACHIRAL ensures the correct impropers are used to define sidechain chirality when HB hydrogen is missing.
- UNIFORMMOVE: the proposed steps for each atom in takestep are uniformly
distributed over a sphere whose radius is adjusted on-the-fly to give the required acceptance
ratio (or fixed if FIXSTEP or FIXBOTH is set).
The default behaviour for backwards compatibility is for the separate ,
 and displacements
of each atom to be scaled uniformly within the current step size range.
and displacements
of each atom to be scaled uniformly within the current step size range.
- UPDATES nup: UPDATES is the number of previous steps saved in the LBFGS routine,
default 4.
- UPDATERIGIDREF: Updates the rigid body reference coordinates after a step has been taken when using
RIGIDINIT. This allows steps to be taken within rigid bodies if required. As unphysical conformations may be introduced
into the rigid bodies this way, HYBRIDMIN should be allowed these to be relaxed.
- USEROT: Include the rigid rotor partition function during a free energy basin-hopping run.
- VGW ljsigma ljepsilon taumaxsg taumaxfg: Specifies use of VGW quantum quenching in place of
classical minimization routines such as LBFGS. ljsigma and ljepsilon are the corresponding Lennard-Jones
parameters that must be specified, and taumaxsg and taumaxfg are the maximum value of ``imaginary'' time
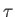 (inverse tempertaure) for the propagation.
The former pertains to the faster ``single-particle'' SP-VGW used for quenching during the MC runs, and the latter for the more accurate
``fully-coupled'' VGW used for the final quenching (analogous to the tight convergence of the LBFGS). A of at least
2.5 is recommended for the SP-VGW and 5.0 for the FC-VGW. A file vgwdata containing the masses (in a.m.u.) of all particles, in order of the location
of their xyz coordinates in ``coords'' must be present (e.g. for a 38 atom Ne cluster, vgwdata will have 38 lines of ``20''). Different
masses are permitted, though the current version allows for only one set of LJ parameters.
(inverse tempertaure) for the propagation.
The former pertains to the faster ``single-particle'' SP-VGW used for quenching during the MC runs, and the latter for the more accurate
``fully-coupled'' VGW used for the final quenching (analogous to the tight convergence of the LBFGS). A of at least
2.5 is recommended for the SP-VGW and 5.0 for the FC-VGW. A file vgwdata containing the masses (in a.m.u.) of all particles, in order of the location
of their xyz coordinates in ``coords'' must be present (e.g. for a 38 atom Ne cluster, vgwdata will have 38 lines of ``20''). Different
masses are permitted, though the current version allows for only one set of LJ parameters.
- VGWCPS on magnitude: Specifies use of contraining potential for SP-VGW (sloppy convergence), as clusters expand during quantum quenching
with decreasing mass. 1 or 0 for on corresponds to on/off,
and magnitude should range from 1 to 1000, with 1 having minimal effect, 1000 being highly constrained. Default value is ``on'', with magnitude 1.
- VGWCPF on magnitude: Same as VGWCPS but for FC-VGW, used for the final, full quenching (tight convergence).
- VGWTOL magnitude: Absolute tolerance parameter for differential equation solver used for VGW quenching. Default value is 0.0001.
For highly quantum or ``stiff'' systems this may need to be increased, while it may be decreased for ``softer'' or less quantum systems to enhance
speed.
- VISITPROP: if specified the Wang-Landau convergence is governed by proportionality of visits to the current value of
the modification factor, and not the histogram flatness criterion [4].
- WELCH
 : specifies a Welch binary
salt potential with the parameters indicated.
: specifies a Welch binary
salt potential with the parameters indicated.
- ZETT1 and ZETT2: specify the Zetterling potentials.
![$\displaystyle \phi(r) = \left(\frac{n}{n-m}\right) \left(\frac{n}{m}\right)^{m/...
... \left(\frac{\sigma}{r}\right)^{n} - \left(\frac{\sigma}{r}\right)^{m}\right].
$](img128.png)
![\includegraphics[width=0.5\textwidth]{PHE.eps}](img151.png)
![$\displaystyle RADIUS=r_e\left[1 + \left(3 n \over 4\pi\sqrt{2}\right)^{1/3}\right], $](img165.png)