[number of sites in rigid body] class [interaction class] site [x] [y] [z] orient [px] [py] [pz] class [interaction class] site [x] [y] [z] orient [px] [py] [pz]where [x] is the x-coordinates of the site in the molecule frame and [px] is the x-component of the angle-axis rotation
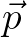 in the molecule frame. For sites with only isotropic interactions, the input for is ignored. For best performance, place the molecule's sites such that the origin corresponds with the molecule's center of mass.
in the molecule frame. For sites with only isotropic interactions, the input for is ignored. For best performance, place the molecule's sites such that the origin corresponds with the molecule's center of mass.
For n-ary mixtures, the syntax is
n-ary [arity] [number fraction of this species] [optional atom symbol] [number of sites in this rigid body] [normal site lines] [number fraction of this species] [number of sites in this rigid body] [normal site lines]The optional atom symbol will cause atoms of different species to have different atom symbols in the output xyz file. Default is `O'.